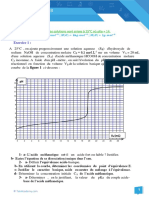
Vous aimerez peut-être aussi
- Terminale S Devoir en Classe N°5 08/04/2006: CHIMIE: L'acide BenzoïqueDocument2 pagesTerminale S Devoir en Classe N°5 08/04/2006: CHIMIE: L'acide BenzoïqueNaouma ChahdPas encore d'évaluation
- Bac-Blanc Ts-Correction-2019-4Document8 pagesBac-Blanc Ts-Correction-2019-4abderrahmane lalaouiPas encore d'évaluation
- acide-carboxylique-inconnuDocument6 pagesacide-carboxylique-inconnuaya.rim.riahiPas encore d'évaluation
- C6Chim - Titrages - Exos - Ph&conduct&color PDFDocument9 pagesC6Chim - Titrages - Exos - Ph&conduct&color PDFAzizElheniPas encore d'évaluation
- Exercices 6Transformations Liées à Des Réactions Acido – Basiques Dans Une Solution AqueuseDocument1 pageExercices 6Transformations Liées à Des Réactions Acido – Basiques Dans Une Solution Aqueuselabiadsihame1Pas encore d'évaluation
- 2023 Asie J2 Exo1 Sujet VitamineC 9ptsDocument5 pages2023 Asie J2 Exo1 Sujet VitamineC 9ptsrabiiyoussef.benaliPas encore d'évaluation
- Résumé 2 Chimie Des SolutionsDocument2 pagesRésumé 2 Chimie Des SolutionsLoubna El JehouariPas encore d'évaluation
- Serie CoronaDocument15 pagesSerie CoronaJoséphine NancassePas encore d'évaluation
- PH SolutionDocument5 pagesPH SolutionBelgasem AsselPas encore d'évaluation
- TP Dosage de L'acide Phosphorique Dans Un Boisson de Coca MPSIDocument3 pagesTP Dosage de L'acide Phosphorique Dans Un Boisson de Coca MPSIsafafars2005Pas encore d'évaluation
- Bac S 2018 Metropole Correction © Exercice IDocument2 pagesBac S 2018 Metropole Correction © Exercice IRemi Laloy100% (1)
- PH Acide FaibleDocument31 pagesPH Acide FaibleAnonymous FikOnlapPas encore d'évaluation
- Ex 1Document6 pagesEx 1teghre chekhne El koryPas encore d'évaluation
- 6129e58d84154reactions Acide Base Solutions Tampons Chimie TermDocument7 pages6129e58d84154reactions Acide Base Solutions Tampons Chimie TermPrince Informaticien GamesPas encore d'évaluation
- Cours Dosage Acide BaseDocument9 pagesCours Dosage Acide BaseBoussetta ZouhaierPas encore d'évaluation
- TleD - CH - L10 - Réactions Acidobasiques. Solutions TamponsDocument21 pagesTleD - CH - L10 - Réactions Acidobasiques. Solutions TamponsHamadi DialloPas encore d'évaluation
- Bac C TG 2022 Maths & PCDocument5 pagesBac C TG 2022 Maths & PCMamadou DioufPas encore d'évaluation
- Cours Dosage Acide Base 3Document8 pagesCours Dosage Acide Base 3Boussetta ZouhaierPas encore d'évaluation
- Série D'exercices - Sciences Physiques Dosage - Bac Math + SC Exp (2014-2015)Document4 pagesSérie D'exercices - Sciences Physiques Dosage - Bac Math + SC Exp (2014-2015)Serigne Abdou NiassePas encore d'évaluation
- Neutralidation Acide Base 2021 2022Document10 pagesNeutralidation Acide Base 2021 2022ZogoPas encore d'évaluation
- C8-Sol Tampon PDFDocument7 pagesC8-Sol Tampon PDFsKiroxPas encore d'évaluation
- Ds Destop CorrecDocument11 pagesDs Destop CorrecDenis BurettePas encore d'évaluation
- Dosage DDocument3 pagesDosage DAzzouze HalaouetPas encore d'évaluation
- 2019 AmNord Exo3 Correction LaitChevre 5ptsDocument3 pages2019 AmNord Exo3 Correction LaitChevre 5ptssaladeaucacaPas encore d'évaluation
- Séance 9Document13 pagesSéance 9SantamitoPas encore d'évaluation
- 2008-09-Antilles-Exo3-Sujet-Titrage-DemiEq-pKa-4ptsDocument2 pages2008-09-Antilles-Exo3-Sujet-Titrage-DemiEq-pKa-4ptsPhysics BdarijaPas encore d'évaluation
- PH Des Solutions Aqueuses 023 0Document10 pagesPH Des Solutions Aqueuses 023 0km.aouadiPas encore d'évaluation
- 2008 09 Antilles Exo3 Sujet Titrage DemiEq Pka 4ptsDocument2 pages2008 09 Antilles Exo3 Sujet Titrage DemiEq Pka 4ptsla physique selon le programme Français100% (1)
- Dosage PHDocument2 pagesDosage PHOumar TraoréPas encore d'évaluation
- Sujetbb 12 Spe 29 JanvDocument6 pagesSujetbb 12 Spe 29 JanvAmza FarelPas encore d'évaluation
- CS Ensa 2020-21Document103 pagesCS Ensa 2020-213freefire 3gamePas encore d'évaluation
- TD Acide Base 24 Fevrier 2016Document10 pagesTD Acide Base 24 Fevrier 2016Tkra G Sil100% (1)
- CocaDocument5 pagesCocatheobromine100% (4)
- Le PH 6. Le Dosage Des Solutions Dacides Et Des Bases Faibles - Theorie - 2014Document15 pagesLe PH 6. Le Dosage Des Solutions Dacides Et Des Bases Faibles - Theorie - 2014RachidaPas encore d'évaluation
- 26 Dosage 3Document3 pages26 Dosage 3Ilyes Ben Jemaa0% (1)
- Lec 15 Les PHDocument12 pagesLec 15 Les PHPierreEustachePas encore d'évaluation
- Chimie Générale4 PDFDocument50 pagesChimie Générale4 PDFTOUREPas encore d'évaluation
- DosageDocument2 pagesDosagePhysique ChimiePas encore d'évaluation
- DosageDocument3 pagesDosageNourhene AbidPas encore d'évaluation
- Alcools 2023 LSLL - WahabdiopDocument2 pagesAlcools 2023 LSLL - WahabdiopMatty DiopPas encore d'évaluation
- Corrige - Examen de Chimie Des Solutions (SMPC - s2) Session Normale-1Document7 pagesCorrige - Examen de Chimie Des Solutions (SMPC - s2) Session Normale-1charki zakariaPas encore d'évaluation
- Exos CH 06Document2 pagesExos CH 06lol testPas encore d'évaluation
- DS2_Tspé_2020-2021-5Document5 pagesDS2_Tspé_2020-2021-5hanaemaaroufPas encore d'évaluation
- TD4 - Réaction Acido BasiqueDocument8 pagesTD4 - Réaction Acido Basiquemelo.lasserrePas encore d'évaluation
- Serie 2 PHDocument3 pagesSerie 2 PHWassim Ben TanfousPas encore d'évaluation
- TP Chimie AnalytiqueDocument13 pagesTP Chimie AnalytiqueFedoua Benamer100% (1)
- Alcools 2023 Lsll-WahabdiopDocument2 pagesAlcools 2023 Lsll-Wahabdioppapa samba sarrPas encore d'évaluation
- Gsa 2bac 1819 Eb1 PPC FRDocument5 pagesGsa 2bac 1819 Eb1 PPC FRIssam ElPas encore d'évaluation
- TransReaAcideBaseExercices 16-17 PDFDocument7 pagesTransReaAcideBaseExercices 16-17 PDFالغزيزال الحسن EL GHZIZAL HassanePas encore d'évaluation
- Fichier Produit 2232Document48 pagesFichier Produit 2232FlorinaPas encore d'évaluation
- DOSAGE COLORIMETRIQUE Et Phmétrique Aspirine CorrigéDocument3 pagesDOSAGE COLORIMETRIQUE Et Phmétrique Aspirine Corrigétdi44617Pas encore d'évaluation
- LycéeBBangangté Chimie TleCD BaccBlanc3 2019 PDFDocument4 pagesLycéeBBangangté Chimie TleCD BaccBlanc3 2019 PDFESSOME ESSOME OLIVIER STEPHANEPas encore d'évaluation
- TD2 ChimieDocument3 pagesTD2 ChimieSAMIA AQNOUCHPas encore d'évaluation
- Chapitre 18Document10 pagesChapitre 18Ayman Ebn taouaitPas encore d'évaluation
- CHAPITRE I - Chimie en SolutionDocument3 pagesCHAPITRE I - Chimie en SolutionNikabou Napo TCHANDIKOUPas encore d'évaluation
- TleD - CH - L11 - Dosage Acido-BasiqueDocument10 pagesTleD - CH - L11 - Dosage Acido-BasiqueHamadi DialloPas encore d'évaluation
- 63c6cf6562315 - Énoncé - Serie18 PH D'une Solution AqueuseDocument3 pages63c6cf6562315 - Énoncé - Serie18 PH D'une Solution AqueuseBen Mabrouk KoussayPas encore d'évaluation
- Exercice Chimie 3Document8 pagesExercice Chimie 3Idris HammouchePas encore d'évaluation
- TD 3ème CEGDocument25 pagesTD 3ème CEGAlain DEMBIPas encore d'évaluation
- Annonce Des AxesDocument4 pagesAnnonce Des AxesOmar BennourPas encore d'évaluation
- Le TravailDocument1 pageLe TravailOmar BennourPas encore d'évaluation
- L'ulcèreDocument3 pagesL'ulcèreOmar BennourPas encore d'évaluation
- SVT Projet SalmaDocument19 pagesSVT Projet SalmaOmar BennourPas encore d'évaluation
- UCAD-ESP - Etude Générale Des Additifs AlimentairesDocument47 pagesUCAD-ESP - Etude Générale Des Additifs AlimentairesMohamed GueyePas encore d'évaluation
- 1as DC II 03 21Document2 pages1as DC II 03 21Abid ElyesPas encore d'évaluation
- Ch11 La Durabilite Des Betons Face Aux Reactions de Gonflements EndogenesDocument12 pagesCh11 La Durabilite Des Betons Face Aux Reactions de Gonflements Endogenesanele_amisPas encore d'évaluation
- Résumé SucrerieDocument5 pagesRésumé SucrerieAbderrahmane HadjadjPas encore d'évaluation
- Désignation Et Nommage Des Câbles Harmonisés - DestockableDocument7 pagesDésignation Et Nommage Des Câbles Harmonisés - Destockablemahmoud akermiPas encore d'évaluation
- Epreuve de Biochimie Session de Mai 2000: Universite Cadi Ayyad Faculte de Medecine Et de Pharmacie MarrakechDocument3 pagesEpreuve de Biochimie Session de Mai 2000: Universite Cadi Ayyad Faculte de Medecine Et de Pharmacie MarrakechBrahim ChahbanePas encore d'évaluation
- Chapitre 3 - Mesure PneumatiqueDocument10 pagesChapitre 3 - Mesure PneumatiqueSmail LebbalPas encore d'évaluation
- Nanomatériaux Et Ses ApplicationsDocument50 pagesNanomatériaux Et Ses Applicationsan hamPas encore d'évaluation
- Atg 3 Facteurs Influencant Atg Suite 2018 2019Document12 pagesAtg 3 Facteurs Influencant Atg Suite 2018 2019Souddi RachidPas encore d'évaluation
- Bio Cell 6Document5 pagesBio Cell 6Khyarhoum BrahimPas encore d'évaluation
- Controle 1 - 25 S1Document2 pagesControle 1 - 25 S1moulay driss100% (1)
- Etancheite en Montagne FasciculeDocument36 pagesEtancheite en Montagne FasciculeSebastien NoguéPas encore d'évaluation
- Chimie 8ème AnnéeDocument14 pagesChimie 8ème AnnéeHama Coulibaly100% (25)
- 1686-Texte de L'article-3417-1-10-20160630Document7 pages1686-Texte de L'article-3417-1-10-20160630IKRAM SanPas encore d'évaluation
- Catalogue Candelis 2019Document17 pagesCatalogue Candelis 2019club Amal Essaouira athlétismePas encore d'évaluation
- TP 1 Microbiologie AlimentaireDocument6 pagesTP 1 Microbiologie AlimentairelarabinouhaPas encore d'évaluation
- LipidesDocument17 pagesLipidesRezagui ImadPas encore d'évaluation
- ITE Coupleurs Et Raccords AutomatiquesDocument46 pagesITE Coupleurs Et Raccords AutomatiquesDiana DianaPas encore d'évaluation
- Cours de Cristallochimie Master IDocument69 pagesCours de Cristallochimie Master IYoussef Laaouje100% (1)
- 1 Les Conducteurs Et CablesDocument5 pages1 Les Conducteurs Et Cables...100% (1)
- Gxv340-390 Manuel Utilisateur 070402Document32 pagesGxv340-390 Manuel Utilisateur 070402math62210Pas encore d'évaluation
- Memoire FinalDocument86 pagesMemoire FinalRajae El-ghadfa100% (2)
- Le RaclageDocument25 pagesLe RaclageImepa S'omyènèPas encore d'évaluation
- 2002 03Document22 pages2002 03rrick_ast8344Pas encore d'évaluation
- Hempathane Topcoat 55210 PDFDocument2 pagesHempathane Topcoat 55210 PDFAhmed HoufPas encore d'évaluation
- Exercices MNF 2 (3) - 075756Document19 pagesExercices MNF 2 (3) - 075756CharlesPas encore d'évaluation
- La Couture Au Fil Des Jours PDFDocument119 pagesLa Couture Au Fil Des Jours PDFLamine86% (7)
- TH2015DeliereLudovic PDFDocument240 pagesTH2015DeliereLudovic PDFmousserPas encore d'évaluation
- Duplex Stainless Steel French PDFDocument64 pagesDuplex Stainless Steel French PDFrachedPas encore d'évaluation
- I Partie CoursDocument162 pagesI Partie CoursPetro DollarsPas encore d'évaluation