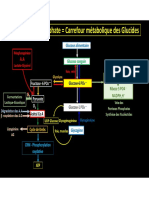
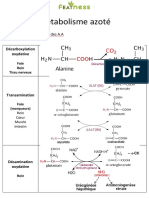
Vous aimerez peut-être aussi
- Southern Blot-Hybridation-SondeDocument51 pagesSouthern Blot-Hybridation-Sondeyouness.khalfaoui100% (4)
- Biosynthèse Des AminoacidesDocument37 pagesBiosynthèse Des AminoacidesImene ImenePas encore d'évaluation
- 2 Metblsme Purines L2 02mars 2021Document12 pages2 Metblsme Purines L2 02mars 2021TETY FREJUS BEUGREPas encore d'évaluation
- Métabolisme Des Acides Aminés Cours de Biochimie 2eme Année Pharmacie DR CHERIETDocument15 pagesMétabolisme Des Acides Aminés Cours de Biochimie 2eme Année Pharmacie DR CHERIETHea FPas encore d'évaluation
- Pharm09 Bioch-Exploration Acides AminesDocument48 pagesPharm09 Bioch-Exploration Acides AminesHicham BenaoumPas encore d'évaluation
- Presentation 112518Document33 pagesPresentation 112518Imene ImenePas encore d'évaluation
- UE7 Annales Interrelations Métab Kilhoffer 2013-16Document9 pagesUE7 Annales Interrelations Métab Kilhoffer 2013-16ElmaPas encore d'évaluation
- Metabolisme Des Acides NucleiquesDocument17 pagesMetabolisme Des Acides Nucleiquesmintchimechak2Pas encore d'évaluation
- Régulation Du Métabolisme Protéique P1Document34 pagesRégulation Du Métabolisme Protéique P1Hicham EddaoudiPas encore d'évaluation
- 01.2 GlycolyseDocument5 pages01.2 Glycolysewassimm31Pas encore d'évaluation
- Chap 11Document18 pagesChap 11glovingimbi62Pas encore d'évaluation
- Métabolisme Des Acides Aminés PDFDocument12 pagesMétabolisme Des Acides Aminés PDFHea FPas encore d'évaluation
- Métabolisme ProtéiqueDocument53 pagesMétabolisme ProtéiqueallaniPas encore d'évaluation
- 18-Mb Des Bases Puriques Et Pyrimidiques2Document22 pages18-Mb Des Bases Puriques Et Pyrimidiques2Hana KouissaPas encore d'évaluation
- Biochimie MicrobienneDocument29 pagesBiochimie Microbiennesino spagoPas encore d'évaluation
- Biochimie. Protéines PDFDocument70 pagesBiochimie. Protéines PDFBrahim Kotto Abdel-azizPas encore d'évaluation
- Biochimie StructuralDocument42 pagesBiochimie StructuralZakariya nadirPas encore d'évaluation
- Vue Générale Du Métabolisme Des GlucidesDocument18 pagesVue Générale Du Métabolisme Des GlucidesKargamel PeyoPas encore d'évaluation
- La NeoglucogeneseDocument2 pagesLa NeoglucogeneseBouazzi IslemmPas encore d'évaluation
- 1 s2.0 S0929693X09740941 MainDocument3 pages1 s2.0 S0929693X09740941 Mainaya prodigePas encore d'évaluation
- 1 M - Tabolisme - Du - GRDocument19 pages1 M - Tabolisme - Du - GRtimPas encore d'évaluation
- Métabolisme Des Amino-Acides: Plan Du CoursDocument29 pagesMétabolisme Des Amino-Acides: Plan Du Courshichem lounisPas encore d'évaluation
- Regulation Du Metabolisme Des ProteinesDocument4 pagesRegulation Du Metabolisme Des ProteinesKamar MakPas encore d'évaluation
- Métabolisme Des Acides AminésDocument7 pagesMétabolisme Des Acides AminésOussama FendaliPas encore d'évaluation
- Métabolisme Des Acides AminésDocument87 pagesMétabolisme Des Acides AminésBlueskyindan100% (1)
- BiochimieDocument42 pagesBiochimieAchwak Belfadel100% (1)
- Biochimie A1Document21 pagesBiochimie A1Joseph YaoPas encore d'évaluation
- Corrige - Biochmeta - sv4 - snp16 - AffanyDocument4 pagesCorrige - Biochmeta - sv4 - snp16 - AffanykombindekanamakanesandraPas encore d'évaluation
- 1 6 Structure Des Acides Amines-1Document13 pages1 6 Structure Des Acides Amines-1Eric OuakaraPas encore d'évaluation
- 08 AcidesAminesL2Document16 pages08 AcidesAminesL2Samiha DiPas encore d'évaluation
- 8 GlycolysisDocument17 pages8 GlycolysisKarim OuzerourouPas encore d'évaluation
- Exploration Acides AminesDocument48 pagesExploration Acides AminesAmiraBenhammouPas encore d'évaluation
- EnzymeDocument8 pagesEnzymefifi fifiPas encore d'évaluation
- QCM Supplementaires Biochimie - PurpanDocument15 pagesQCM Supplementaires Biochimie - PurpanMidouri Djaffer67% (3)
- Catabolisme Des Acides Aminés CoursDocument24 pagesCatabolisme Des Acides Aminés Courslaila hammouPas encore d'évaluation
- Catabolisme Des Lipides Complexes L3 BiochimieDocument22 pagesCatabolisme Des Lipides Complexes L3 Biochimiebiochimie L3Pas encore d'évaluation
- 3 - Metabolisme Des ProtidesDocument14 pages3 - Metabolisme Des ProtidesPascal GadedjissoPas encore d'évaluation
- Acides Amin S Et Cycle Ur eDocument44 pagesAcides Amin S Et Cycle Ur eDominique MEYERPas encore d'évaluation
- Biochimie - GlucidesDocument51 pagesBiochimie - GlucidesmimiPas encore d'évaluation
- Cours 8 A GLYCOLYSEDocument15 pagesCours 8 A GLYCOLYSELina BernoussiPas encore d'évaluation
- Hyperoh 3Document66 pagesHyperoh 3Lilo LiliPas encore d'évaluation
- Voie Des Pentoses PhosphatesDocument26 pagesVoie Des Pentoses Phosphatescep100% (1)
- Métabolisme Protéines Fiche PDFDocument4 pagesMétabolisme Protéines Fiche PDFNaïs LABBADIPas encore d'évaluation
- 5 - Catabolisme Des AADocument14 pages5 - Catabolisme Des AAMey ZPas encore d'évaluation
- Digestion Des Disaccharides + Absorption 2021 ÉtudiantsDocument37 pagesDigestion Des Disaccharides + Absorption 2021 ÉtudiantsbrahmiPas encore d'évaluation
- 1 - Acides AminesDocument48 pages1 - Acides AminesHadjer SeddikiPas encore d'évaluation
- LESONA SVT 1ere DDocument124 pagesLESONA SVT 1ere DAndrew RanPas encore d'évaluation
- Métabolisme Des NucléotidesDocument44 pagesMétabolisme Des NucléotidesflowershengPas encore d'évaluation
- Les ProtéinesDocument11 pagesLes ProtéinesFéz EyPas encore d'évaluation
- Cycle de L'uree...Document21 pagesCycle de L'uree...hanane ElachhabPas encore d'évaluation
- 03-Glycolyse 2021Document7 pages03-Glycolyse 2021Mery Sage100% (1)
- 01.4 Voie Des Pentoses PhosphatesDocument5 pages01.4 Voie Des Pentoses Phosphateswassimm31Pas encore d'évaluation
- Métabolisme Des Acides AminésDocument40 pagesMétabolisme Des Acides AminéscepPas encore d'évaluation
- Catabolisme Des GlucidesDocument11 pagesCatabolisme Des Glucidesevident 2014Pas encore d'évaluation
- BiomaDocument99 pagesBiomaFarid KhalloukiPas encore d'évaluation
- Glossaire de BiochimieDocument26 pagesGlossaire de BiochimieBoris SosnovPas encore d'évaluation
- Les Acides AminesDocument3 pagesLes Acides AminesCoo Kie100% (1)
- A/ Glycolyse (Ou Voie d'Embden-Meyerhof) : A) Les Différentes Étapes de La GlycolyseDocument6 pagesA/ Glycolyse (Ou Voie d'Embden-Meyerhof) : A) Les Différentes Étapes de La GlycolysemaryPas encore d'évaluation
- Les Aminoacidopathies PDFDocument13 pagesLes Aminoacidopathies PDFAmira AmraouiPas encore d'évaluation
- Cours - 02 - Nature Chimiques Du Matériel GénétiqueDocument34 pagesCours - 02 - Nature Chimiques Du Matériel Génétiquejames deyPas encore d'évaluation
- La Transcription l3 Microbiologie 2022Document8 pagesLa Transcription l3 Microbiologie 2022rou röuPas encore d'évaluation
- Cours Lipides 20 21Document16 pagesCours Lipides 20 21oumayma salihiPas encore d'évaluation
- La Transcription de l'ADN 1Document23 pagesLa Transcription de l'ADN 1Ismail ZitouniPas encore d'évaluation
- TD Peptitides Et Protéines 2000-2001Document3 pagesTD Peptitides Et Protéines 2000-2001Ikram FerPas encore d'évaluation
- UntitledtdugogrDocument3 pagesUntitledtdugogrLoubna MichaPas encore d'évaluation
- Macro NutrientsDocument6 pagesMacro NutrientsTian HePas encore d'évaluation
- Chuyen Hoa ChungDocument20 pagesChuyen Hoa ChungYêu QuáiiPas encore d'évaluation
- Année 2021-2022-EPSS - Exo Entrainement - 07/2022Document2 pagesAnnée 2021-2022-EPSS - Exo Entrainement - 07/2022Ivan AdouPas encore d'évaluation
- Metabolisme Des LipidesDocument97 pagesMetabolisme Des LipidesFikra PapPas encore d'évaluation
- Cours - Biochimie Structurale - Lipides - CAG - UNA - 2019 - CopieDocument83 pagesCours - Biochimie Structurale - Lipides - CAG - UNA - 2019 - CopieStellaPas encore d'évaluation
- S1 TD2 Protides PDFDocument8 pagesS1 TD2 Protides PDFmartinmune67% (3)
- TRAnscriptionDocument16 pagesTRAnscriptionmaysounPas encore d'évaluation
- Cours1 GlucidesDocument49 pagesCours1 GlucidesLinda KoundziPas encore d'évaluation
- CHEIDIDocument4 pagesCHEIDIcheilla nandjioPas encore d'évaluation
- TD 2-bcfDocument17 pagesTD 2-bcfFatïma ZohraPas encore d'évaluation
- TD LipidesDocument5 pagesTD LipidesWissam Tiza100% (1)
- PCEM 1 Examen Blanc 2019Document7 pagesPCEM 1 Examen Blanc 2019Mou Medib100% (1)
- SICOLAY 1035 PondeusesDocument1 pageSICOLAY 1035 PondeusesMenna ElsayedPas encore d'évaluation
- Lipo ProteineDocument11 pagesLipo ProteineAly YalcouyéPas encore d'évaluation
- Biologie Moleculaire MunirDocument42 pagesBiologie Moleculaire MunirOuu SsaamaPas encore d'évaluation
- CoenzymeDocument11 pagesCoenzymemido benPas encore d'évaluation
- Chapitre II La Réplication de LADNDocument37 pagesChapitre II La Réplication de LADNfekhar adlenPas encore d'évaluation
- MacronutrimentDocument1 pageMacronutrimentRafik BenhendaPas encore d'évaluation
- Structure Des Membranes Lipidiques 1Document51 pagesStructure Des Membranes Lipidiques 1Abdellah EL AnsariPas encore d'évaluation
- Genie Genetique Chapitre 3Document5 pagesGenie Genetique Chapitre 3Ñoor ĒllîzzaPas encore d'évaluation
- Tdbiochimie - Solutions ConvertiDocument18 pagesTdbiochimie - Solutions ConvertinantenainaarthinaPas encore d'évaluation
- 1.3. La Structure Des MembranesDocument9 pages1.3. La Structure Des MembranesAstrid Gentelle Ngatchou TchuisseuPas encore d'évaluation
- TD 3 de BiochimieDocument5 pagesTD 3 de BiochimieRich HoulrichPas encore d'évaluation