Vous aimerez peut-être aussi
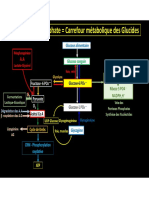
- NéoglucogénèseDocument1 pageNéoglucogénèseHadrien De GreefPas encore d'évaluation
- Image Code Couleur TubesDocument2 pagesImage Code Couleur TubesFairouz Tou100% (2)
- 2010 Rouen Dubus CoursBiopchimieAn2P4 PDFDocument27 pages2010 Rouen Dubus CoursBiopchimieAn2P4 PDFOumaima BelPas encore d'évaluation
- Pr អ៊ុង ចាន់នី y15-18Document94 pagesPr អ៊ុង ចាន់នី y15-18varyvira6677Pas encore d'évaluation
- 13.mtabolisme Des Acides Gras.Document41 pages13.mtabolisme Des Acides Gras.paul destinPas encore d'évaluation
- Métabolisme Des Amino-Acides: Plan Du CoursDocument29 pagesMétabolisme Des Amino-Acides: Plan Du Courshichem lounisPas encore d'évaluation
- Protéine RésuméDocument10 pagesProtéine RésuméShanone FatimaPas encore d'évaluation
- Biochimie. Protéines PDFDocument70 pagesBiochimie. Protéines PDFBrahim Kotto Abdel-azizPas encore d'évaluation
- Liste Des Ciq Et Parametres Au Poste de BiochimieDocument3 pagesListe Des Ciq Et Parametres Au Poste de BiochimieIsabelle Usclade SophrologuePas encore d'évaluation
- METABOLISME LIPIDIQUE DFGSP-2 ERdeMDocument10 pagesMETABOLISME LIPIDIQUE DFGSP-2 ERdeMsikaokoumassouPas encore d'évaluation
- Digestion Pancréatique Protéines 2021 ÉtudiantsDocument48 pagesDigestion Pancréatique Protéines 2021 ÉtudiantsbrahmiPas encore d'évaluation
- Métabolisme Des Acides AminésDocument87 pagesMétabolisme Des Acides AminésBlueskyindan100% (1)
- Biosynthèse Des AminoacidesDocument37 pagesBiosynthèse Des AminoacidesImene ImenePas encore d'évaluation
- Vue Générale Du Métabolisme Des GlucidesDocument18 pagesVue Générale Du Métabolisme Des GlucidesKargamel PeyoPas encore d'évaluation
- Biochimie Rénale (Important)Document7 pagesBiochimie Rénale (Important)Idrissou FmsbPas encore d'évaluation
- Catabolisme Des Acides Aminés CoursDocument24 pagesCatabolisme Des Acides Aminés Courslaila hammouPas encore d'évaluation
- Cycle UreeDocument1 pageCycle UreeJordan SassoPas encore d'évaluation
- Cours Bioch Métab Version CP 2 Agro IAV Partie 2Document135 pagesCours Bioch Métab Version CP 2 Agro IAV Partie 2Your fav Aya100% (1)
- Corrige Type Du TD 9 1-Converti PDFDocument4 pagesCorrige Type Du TD 9 1-Converti PDFANDRE DESTAIN MBEZELE ZEPas encore d'évaluation
- Planchage de Biosynthese Des Acides Amines Final (1) .PPTX Version 1Document32 pagesPlanchage de Biosynthese Des Acides Amines Final (1) .PPTX Version 1Imene ImenePas encore d'évaluation
- C. Biochimie III. Lipides (III.1. Métabolisme Des Acides Gras)Document41 pagesC. Biochimie III. Lipides (III.1. Métabolisme Des Acides Gras)Rania HamdiPas encore d'évaluation
- P2oxydation Corps GrasDocument19 pagesP2oxydation Corps GrasbelhadiePas encore d'évaluation
- Catabolisme Des Aa - Vue Generale - CompressedDocument43 pagesCatabolisme Des Aa - Vue Generale - CompressedRaouf ChikhiPas encore d'évaluation
- Tableau EnzymesDocument2 pagesTableau Enzymesclemcg78Pas encore d'évaluation
- Régulation Du Métabolisme Protéique P1Document34 pagesRégulation Du Métabolisme Protéique P1Hicham EddaoudiPas encore d'évaluation
- Prépabio s1 DocumentsDocument12 pagesPrépabio s1 DocumentsDavid LemenagerPas encore d'évaluation
- Métab. Des AA-VF-22Document89 pagesMétab. Des AA-VF-22s22g9ndkwjPas encore d'évaluation
- Catabolisme Des Acides AminesDocument24 pagesCatabolisme Des Acides AminesBoutheina HafPas encore d'évaluation
- Tableau EnzymesDocument2 pagesTableau Enzymesclemcg78Pas encore d'évaluation
- Cours 2 Nutrition Thérapeutique AvancéDocument20 pagesCours 2 Nutrition Thérapeutique Avancéamina imenePas encore d'évaluation
- BiochimieDocument42 pagesBiochimieAchwak Belfadel100% (1)
- CoursBiopchimie PDFDocument14 pagesCoursBiopchimie PDFLILIA HAMROUNIPas encore d'évaluation
- Metabolisme LipidesDocument49 pagesMetabolisme LipidesMohamed Ali MtibaaPas encore d'évaluation
- Examen 2015 Ratt CorrigeDocument9 pagesExamen 2015 Ratt Corrigefawzi badiPas encore d'évaluation
- Cours 3 Physiologie de L'alimentation Et MetabolismeDocument50 pagesCours 3 Physiologie de L'alimentation Et Metabolismedido poloPas encore d'évaluation
- Flyer MT Reactifs Genrui Pour Analyseurs Biochimie 032021 FR WebDocument2 pagesFlyer MT Reactifs Genrui Pour Analyseurs Biochimie 032021 FR Webamor kermayaPas encore d'évaluation
- Enzyme Biochimie - TableauxDocument7 pagesEnzyme Biochimie - TableauxMed BlhhPas encore d'évaluation
- Burkina 2014 SVT Serie D 1er Tour Sujet 1 RempDocument4 pagesBurkina 2014 SVT Serie D 1er Tour Sujet 1 RempAbané Jean davidPas encore d'évaluation
- 08 AcidesAminesL2Document16 pages08 AcidesAminesL2Samiha DiPas encore d'évaluation
- Examen Biologique AbreviationDocument2 pagesExamen Biologique AbreviationImadPas encore d'évaluation
- Metabolisme LipidesDocument49 pagesMetabolisme LipidespartiraretirapasPas encore d'évaluation
- Structure Des Lipides EF 2016-17 PACES UE1 - Cours 1 - 2 Et 3Document29 pagesStructure Des Lipides EF 2016-17 PACES UE1 - Cours 1 - 2 Et 3Yousra INDIAPas encore d'évaluation
- Les Lipides Et Dérivés1Document66 pagesLes Lipides Et Dérivés1SALOUAPas encore d'évaluation
- Cycle de Krebs Ou de L Acide CitriqueDocument13 pagesCycle de Krebs Ou de L Acide CitriqueJean-Loïc BauchetPas encore d'évaluation
- 1ere Partie Bio Tse 12212Document17 pages1ere Partie Bio Tse 12212Sawoulou celest DopavoguiPas encore d'évaluation
- Biochimie StructuralDocument42 pagesBiochimie StructuralZakariya nadirPas encore d'évaluation
- Fiche Prises de Sang ESIDocument1 pageFiche Prises de Sang ESISibylle TnnPas encore d'évaluation
- Guide Simplifié A5 Juin 2022 3Document2 pagesGuide Simplifié A5 Juin 2022 3KACH 4Pas encore d'évaluation
- Cours 4 Rôle Des Composants Membranaires Dans La Signalisation CellulaireDocument15 pagesCours 4 Rôle Des Composants Membranaires Dans La Signalisation CellulaireredPas encore d'évaluation
- Le Cycle de KrebsDocument19 pagesLe Cycle de KrebsLandry RimbaybarPas encore d'évaluation
- Regulation Du Metabolisme Des ProteinesDocument4 pagesRegulation Du Metabolisme Des ProteinesKamar MakPas encore d'évaluation
- Presentation 112518Document33 pagesPresentation 112518Imene ImenePas encore d'évaluation
- Biosynthése Des Lipidsynt10Document9 pagesBiosynthése Des Lipidsynt10mhichePas encore d'évaluation
- Les Acides AminesDocument7 pagesLes Acides Aminesbouguidima_sanaPas encore d'évaluation
- Métabolisme Lipides PDFDocument8 pagesMétabolisme Lipides PDFNaïs LABBADIPas encore d'évaluation
- Production de L'énergie Par OxydationDocument2 pagesProduction de L'énergie Par OxydationRamzi BoulahmiPas encore d'évaluation
- Pharm09 Bioch-Exploration Acides AminesDocument48 pagesPharm09 Bioch-Exploration Acides AminesHicham BenaoumPas encore d'évaluation
- Ob - 4e7114 - Expose Sur La Natation Liztout 2014Document20 pagesOb - 4e7114 - Expose Sur La Natation Liztout 2014Lilo LiliPas encore d'évaluation
- Necrosis Factor), Les Neuromédiateurs, Le Stress Cer-: Interleukine 1Document2 pagesNecrosis Factor), Les Neuromédiateurs, Le Stress Cer-: Interleukine 1Lilo LiliPas encore d'évaluation
- Code Du SportDocument686 pagesCode Du SportLilo LiliPas encore d'évaluation
- Manuel Alimentation Et Grossesse SSN 2018Document67 pagesManuel Alimentation Et Grossesse SSN 2018Lilo LiliPas encore d'évaluation
- Assurer Un Apport Alimentaire ÉquilibréDocument1 pageAssurer Un Apport Alimentaire ÉquilibréLilo LiliPas encore d'évaluation
- ALIMENTS RICHES EN ZINC PDF Tableau 1Document3 pagesALIMENTS RICHES EN ZINC PDF Tableau 1Lilo LiliPas encore d'évaluation
- Les MesuresDocument2 pagesLes MesuresLilo LiliPas encore d'évaluation
- Les Mesures HygiénodiététiquesDocument1 pageLes Mesures HygiénodiététiquesLilo LiliPas encore d'évaluation
- Pour Rappel, Une Glycémie Normale Se Situe Entre 70 Et 100 Milligrammes Par DécilitreDocument1 pagePour Rappel, Une Glycémie Normale Se Situe Entre 70 Et 100 Milligrammes Par DécilitreLilo LiliPas encore d'évaluation
- Untitled DocumentDocument2 pagesUntitled DocumentLilo LiliPas encore d'évaluation
- Anatomo Physio FoieDocument7 pagesAnatomo Physio FoieDjallal HassaniPas encore d'évaluation
- Grossesse Extra-Utérine À Madagascar - 107 ObservationsDocument4 pagesGrossesse Extra-Utérine À Madagascar - 107 ObservationsMahefa Serge RakotozafyPas encore d'évaluation
- DéshydratationDocument14 pagesDéshydratationmessi000013Pas encore d'évaluation
- Eval CE1 Corrigés Juin19Document7 pagesEval CE1 Corrigés Juin19mbiteyene velorPas encore d'évaluation
- Filarioses Lymphatiques PH2 2020Document52 pagesFilarioses Lymphatiques PH2 2020mkdqhp8rwdPas encore d'évaluation
- Avc PDFDocument87 pagesAvc PDFThiziri ThiziriPas encore d'évaluation
- MCCPDocument14 pagesMCCPLahcen BoulahcenPas encore d'évaluation
- Fiche de Renseignement VaccinDocument2 pagesFiche de Renseignement Vaccinghizlane berradaPas encore d'évaluation
- 10 PRO - EXP3 - S10 - G2 - Cours - 06-11 JuinDocument267 pages10 PRO - EXP3 - S10 - G2 - Cours - 06-11 JuinwalidPas encore d'évaluation
- PROGRAMME COMPLET 2eme Annee 2011-2012Document27 pagesPROGRAMME COMPLET 2eme Annee 2011-2012Sid Ali ChinwiPas encore d'évaluation
- Evaluation 102Document4 pagesEvaluation 102Cristian JoianPas encore d'évaluation
- HorlogeDocument8 pagesHorlogeMeryam benyahiaPas encore d'évaluation
- Pancréatite Lithiasique 2018Document13 pagesPancréatite Lithiasique 2018ElbordjiPas encore d'évaluation
- Complications Des Stimulateurs CardiaquesDocument4 pagesComplications Des Stimulateurs CardiaquesdrlauriPas encore d'évaluation
- Contrat D'Accueil Creche: Table Des MatièresDocument8 pagesContrat D'Accueil Creche: Table Des MatièresLila RzinPas encore d'évaluation
- 1°spe Fiche Bilan Immuno C2Document2 pages1°spe Fiche Bilan Immuno C2Nathan VericelPas encore d'évaluation
- Conservation Des AlimentsDocument28 pagesConservation Des AlimentsMel ĀSsîillêm100% (2)
- Carnet de Stage SUSI: October 2015Document65 pagesCarnet de Stage SUSI: October 2015Sanaa AbidPas encore d'évaluation
- Embryo Et DVP Cranio-Faciale 1Document2 pagesEmbryo Et DVP Cranio-Faciale 1Kadiri soumayaPas encore d'évaluation
- NCM 104 - Soins Infirmiers en Santé Communautaire 1Document12 pagesNCM 104 - Soins Infirmiers en Santé Communautaire 1ScribdTranslationsPas encore d'évaluation
- Cours de TrachomeDocument18 pagesCours de TrachomeSylvert DjéranéPas encore d'évaluation
- Fadwa Pfe QuestionnaireDocument5 pagesFadwa Pfe QuestionnaireFadwa AbaidiPas encore d'évaluation
- Tableau Urgences Dentaires BretagneDocument1 pageTableau Urgences Dentaires BretagnebleuPDFPas encore d'évaluation
- 268 Ex 266 CalciumDocument6 pages268 Ex 266 CalciumHmaied BaratliPas encore d'évaluation
- La Trisomie 12 Est Une Anomalie Chromosomique Rare Caractérisée Par La Présence DDocument2 pagesLa Trisomie 12 Est Une Anomalie Chromosomique Rare Caractérisée Par La Présence DMouhammed DIOPPas encore d'évaluation
- IPID-0181 - Odalys-vacances-Annulation interruption-QL7 - CovidDocument2 pagesIPID-0181 - Odalys-vacances-Annulation interruption-QL7 - CovidGENETPas encore d'évaluation
- Thrombose Veineuse Profonde2Document67 pagesThrombose Veineuse Profonde2Serigne Sohibou GayePas encore d'évaluation
- JournalMedecineAntiAge Septembre 2019Document16 pagesJournalMedecineAntiAge Septembre 2019djonoumaPas encore d'évaluation
- QCM MICROBIODocument3 pagesQCM MICROBIOResidanat 2023Pas encore d'évaluation
- 01 Infection Bronchopulmonaire de L'adulteDocument63 pages01 Infection Bronchopulmonaire de L'adulteMichelle DazsPas encore d'évaluation