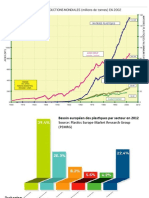
Vous aimerez peut-être aussi
- Chapitre I. Structure Des Polymeres Et Leurs ClassificationsDocument8 pagesChapitre I. Structure Des Polymeres Et Leurs ClassificationsJewel SlamPas encore d'évaluation
- Ero Ds c03Document2 pagesEro Ds c03Nina HanPas encore d'évaluation
- Métal Amorphe: Le verre métallique mince du futur ressemble à du papier d'aluminium, mais essayez de le déchirer, ou voyez si vous pouvez le couper, de toute votre puissance, pas de chanceD'EverandMétal Amorphe: Le verre métallique mince du futur ressemble à du papier d'aluminium, mais essayez de le déchirer, ou voyez si vous pouvez le couper, de toute votre puissance, pas de chancePas encore d'évaluation
- TD1 2Document4 pagesTD1 2Oussama El BouadiPas encore d'évaluation
- Chap. 1-PolymèresDocument5 pagesChap. 1-PolymèresBénédicte & Christophe MULLERPas encore d'évaluation
- Des Applications Sur Les PolymèresDocument2 pagesDes Applications Sur Les PolymèresmortadaPas encore d'évaluation
- Fascicule2022 2023Document18 pagesFascicule2022 2023Lucien NegloPas encore d'évaluation
- Correction TD 2Document4 pagesCorrection TD 2mahmoud bensassiPas encore d'évaluation
- Cours PolymèresDocument17 pagesCours PolymèresJhee raaPas encore d'évaluation
- TD Polymère 1Document1 pageTD Polymère 1mostafa bounabPas encore d'évaluation
- Chimie Des Polymeres Exercices Et Problemes Corriges 2 Ed - SommaireDocument22 pagesChimie Des Polymeres Exercices Et Problemes Corriges 2 Ed - Sommairezina allaouaPas encore d'évaluation
- Iutmosel Polymeres 2012 CHDocument2 pagesIutmosel Polymeres 2012 CHAbdallah AmmarPas encore d'évaluation
- Chapitre II Polymérisation RadicalaireDocument10 pagesChapitre II Polymérisation RadicalaireKherfi CherifaPas encore d'évaluation
- Série 01Document2 pagesSérie 01ibend376Pas encore d'évaluation
- LPro Polym PDFDocument23 pagesLPro Polym PDFRafik Dra100% (1)
- (WWW - Clubetudiants.ma) - Cours de Thermodynamique Nov 2015Document135 pages(WWW - Clubetudiants.ma) - Cours de Thermodynamique Nov 2015Rondello99Pas encore d'évaluation
- Chap1 CristallographieDocument18 pagesChap1 CristallographiezakariaePas encore d'évaluation
- TD 2010 PlastiquesDocument2 pagesTD 2010 PlastiquesmortadaPas encore d'évaluation
- M14 - TD N°2: Spectrométrie Infra Rouge: Master (PACQ)Document2 pagesM14 - TD N°2: Spectrométrie Infra Rouge: Master (PACQ)Imane MouamrPas encore d'évaluation
- Chimie Théorique TD1Document2 pagesChimie Théorique TD1thegthed0% (1)
- Chapitre 3 - STDocument16 pagesChapitre 3 - STDeghboudj SamirPas encore d'évaluation
- Emd1 Phystat 2017Document4 pagesEmd1 Phystat 2017zakari3yaePas encore d'évaluation
- Theorie de La Fonctionnelle de DensiteDocument15 pagesTheorie de La Fonctionnelle de DensiteMouniaPas encore d'évaluation
- Chim430 Organometallique Cours PDFDocument144 pagesChim430 Organometallique Cours PDFAmel AyadiPas encore d'évaluation
- Mécatronique en Partant de RienDocument18 pagesMécatronique en Partant de RienAbdirachid Ali100% (1)
- SupraDocument54 pagesSuprajacques-rene-eone-6805Pas encore d'évaluation
- Courbesintensite 2015 CoursDocument30 pagesCourbesintensite 2015 CourscherniPas encore d'évaluation
- AlcèneDocument6 pagesAlcènechristophe100% (1)
- TD 1 GMP 2023-2024Document4 pagesTD 1 GMP 2023-2024mennanesalaheddinePas encore d'évaluation
- Programme D'agregation Physique ChimieDocument3 pagesProgramme D'agregation Physique ChimieAdilPas encore d'évaluation
- Théorie de La Fonctionnelle de La Densité - Iramis - CEADocument35 pagesThéorie de La Fonctionnelle de La Densité - Iramis - CEAbmalki68Pas encore d'évaluation
- EF1 Corrige Chimie3 19012016Document5 pagesEF1 Corrige Chimie3 19012016martin souman moulsiaPas encore d'évaluation
- Structure Electronique Des MoleculesDocument17 pagesStructure Electronique Des MoleculesFatima EL AROUSSIPas encore d'évaluation
- TD 02Document3 pagesTD 02yousraghanem23Pas encore d'évaluation
- Ero Ex c02Document3 pagesEro Ex c02maino100% (1)
- Cristallochimie 2001Document227 pagesCristallochimie 2001Youssef NajihPas encore d'évaluation
- TP de Cinetique Chimique L2 Polytech - 080923Document4 pagesTP de Cinetique Chimique L2 Polytech - 080923Alain Ditend100% (1)
- Ato ExercicesDocument11 pagesAto ExerciceshhedfiPas encore d'évaluation
- Master2 MFTDocument28 pagesMaster2 MFTbokpedidier1Pas encore d'évaluation
- TD Polymere TcaDocument6 pagesTD Polymere TcaBihina HamanPas encore d'évaluation
- Chapitre 1 de Synthèse (PolycondensationDocument10 pagesChapitre 1 de Synthèse (PolycondensationHana Salah AiechPas encore d'évaluation
- Cours Degradation Polymere N°3Document22 pagesCours Degradation Polymere N°3Ouahes HoudaifaPas encore d'évaluation
- Axlou Toth Pour L'innovation: Cours de Renforcement Ou À Domicile Maths-PC-SVT: 78.192.84.64-78.151.34.44Document3 pagesAxlou Toth Pour L'innovation: Cours de Renforcement Ou À Domicile Maths-PC-SVT: 78.192.84.64-78.151.34.44yayaPas encore d'évaluation
- Chapitre 1Document26 pagesChapitre 1HindPas encore d'évaluation
- Math IV Les Fonctions SpécialesDocument55 pagesMath IV Les Fonctions SpécialesEl-Kaber HachemPas encore d'évaluation
- Cours Chimie Organique SMP S3Document68 pagesCours Chimie Organique SMP S3MOHAMMED ZAKARIA BAALI100% (1)
- TD 2-Cristallo 2019Document1 pageTD 2-Cristallo 2019héma tologiePas encore d'évaluation
- Chrono Potent I Om Ét RieDocument33 pagesChrono Potent I Om Ét RieIsmail HisashiPas encore d'évaluation
- TD Cinétique Chimique Série N°1 CorrectionDocument6 pagesTD Cinétique Chimique Série N°1 CorrectionadnanPas encore d'évaluation
- TD1 L3S6 - 20-21Document4 pagesTD1 L3S6 - 20-21Ilias aatikPas encore d'évaluation
- TD 1 - MQ - MasterDocument2 pagesTD 1 - MQ - MasterWalid Zizi100% (1)
- Cours de Physique StatistiqueDocument171 pagesCours de Physique StatistiqueDaoud El CaidPas encore d'évaluation
- CM Thermo Générale L3 U-ManDocument176 pagesCM Thermo Générale L3 U-ManHarvey SpecterPas encore d'évaluation
- Electricité 3 TD1 ChaibDocument32 pagesElectricité 3 TD1 ChaibDaoud El CaidPas encore d'évaluation
- TD MagnetismeDocument2 pagesTD MagnetismeAyoub BPas encore d'évaluation
- Thermo Stat PBDocument61 pagesThermo Stat PBabdelkanPas encore d'évaluation
- Rappels Thermo Des Solutions MacromoléculairesDocument4 pagesRappels Thermo Des Solutions MacromoléculairesVincent ImadPas encore d'évaluation
- ELJouhariTheorie Des GroupesDocument22 pagesELJouhariTheorie Des GroupesYc YacinePas encore d'évaluation
- Poly RupDocument103 pagesPoly RupIssam HebbouchePas encore d'évaluation
- Materiaux CoursDocument40 pagesMateriaux CoursExode Christ GuiellePas encore d'évaluation
- Contribution A La Caracterisation Non Destructive de Beton Arme Par La Methode Des UltrasonsDocument53 pagesContribution A La Caracterisation Non Destructive de Beton Arme Par La Methode Des UltrasonsSAIB BRAHIMPas encore d'évaluation
- Critères de RuptureDocument6 pagesCritères de RuptureOumaima KehilPas encore d'évaluation
- Batiment Bois Parasismiques FDocument25 pagesBatiment Bois Parasismiques FPala2014Pas encore d'évaluation
- Défaillances Et Loi D'usure PDFDocument3 pagesDéfaillances Et Loi D'usure PDFmohamedPas encore d'évaluation
- G P - MM - WatermarkDocument72 pagesG P - MM - WatermarkMazighi SaadPas encore d'évaluation
- Materiaux de Construction Chap 1Document13 pagesMateriaux de Construction Chap 1YounessElKhattabiPas encore d'évaluation
- RapportDocument40 pagesRapportRayen AbidiPas encore d'évaluation
- Code - Aster: Éléments Finis Traitant La Quasi-IncompressibilitéDocument18 pagesCode - Aster: Éléments Finis Traitant La Quasi-IncompressibilitéFongho Eric SinclairPas encore d'évaluation
- Examen de Structures en PlasticitéDocument5 pagesExamen de Structures en PlasticitéUrsula Joyce Merveilles Pettang Nana100% (1)
- Procédés de Mise en Forme Par Déformation PlastiqueDocument2 pagesProcédés de Mise en Forme Par Déformation PlastiqueHamada HamadaPas encore d'évaluation
- MDC 1chp 1 GeneraliDocument7 pagesMDC 1chp 1 GeneraliSERGIOPas encore d'évaluation
- Principe Du ForgeageDocument22 pagesPrincipe Du ForgeageHamada HamadaPas encore d'évaluation
- Chapitre 33 Structure Comportement Des PolymeresDocument21 pagesChapitre 33 Structure Comportement Des PolymeresCHERIFPas encore d'évaluation
- Résilience TénacitéDocument8 pagesRésilience TénacitéIssaoui MansourPas encore d'évaluation
- SIA260 FDocument44 pagesSIA260 FIris GibourgPas encore d'évaluation
- Laminage À Chaud, Théorie Du LaminageDocument23 pagesLaminage À Chaud, Théorie Du LaminageMOHAMEDPas encore d'évaluation
- TD-Procédés de Fab° MécDocument12 pagesTD-Procédés de Fab° MécHAMZAPas encore d'évaluation
- Sofiane Achache 2016TROY0002Document152 pagesSofiane Achache 2016TROY0002achraf zegnaniPas encore d'évaluation
- Analyse Limite 2021Document40 pagesAnalyse Limite 2021Fisso Ben BenPas encore d'évaluation
- Memoire HDR Franck ToussaintDocument153 pagesMemoire HDR Franck Toussaintbilal.lenjasse.23Pas encore d'évaluation
- 4-Traction-Compression CompaisonDocument13 pages4-Traction-Compression CompaisonIlu SionPas encore d'évaluation
- Biomécanique de L'osDocument17 pagesBiomécanique de L'osaminePas encore d'évaluation
- Memoire FinaleDocument67 pagesMemoire FinaleOint MIMBPas encore d'évaluation
- Le Seisme Et Les Constructions en Métal Et en BoisDocument32 pagesLe Seisme Et Les Constructions en Métal Et en BoislhabsPas encore d'évaluation
- PP AbaqusDocument4 pagesPP Abaqushamza dahbiPas encore d'évaluation
- Chapitre III PlasticitéDocument4 pagesChapitre III PlasticitéskayrooPas encore d'évaluation
- Test Plast 2019-05-23 RepDocument2 pagesTest Plast 2019-05-23 RepjeromedeservignyPas encore d'évaluation
- Krea - Notice TechniqueDocument50 pagesKrea - Notice Techniquesbaia aminePas encore d'évaluation
- Secrets ancestraux d'un maître guérisseur: Un sceptique occidental, un maître oriental et les plus grands secrets de la vieD'EverandSecrets ancestraux d'un maître guérisseur: Un sceptique occidental, un maître oriental et les plus grands secrets de la vieÉvaluation : 5 sur 5 étoiles5/5 (2)
- L'Art de la guerre: Traité de stratégie en 13 chapitres (texte intégral)D'EverandL'Art de la guerre: Traité de stratégie en 13 chapitres (texte intégral)Évaluation : 4 sur 5 étoiles4/5 (3032)
- La vie des abeilles: Prix Nobel de littératureD'EverandLa vie des abeilles: Prix Nobel de littératureÉvaluation : 4 sur 5 étoiles4/5 (41)
- Géobiologie de l'habitat et Géobiologie sacrée: Pour un lieu sainD'EverandGéobiologie de l'habitat et Géobiologie sacrée: Pour un lieu sainÉvaluation : 4.5 sur 5 étoiles4.5/5 (2)
- Manuel pour les débutants Fabriquez des savons naturelsD'EverandManuel pour les débutants Fabriquez des savons naturelsÉvaluation : 3 sur 5 étoiles3/5 (2)
- Améliorer votre mémoire: Un Guide pour l'augmentation de la puissance du cerveau, utilisant des techniques et méthodesD'EverandAméliorer votre mémoire: Un Guide pour l'augmentation de la puissance du cerveau, utilisant des techniques et méthodesÉvaluation : 5 sur 5 étoiles5/5 (2)
- Technologie automobile: Les Grands Articles d'UniversalisD'EverandTechnologie automobile: Les Grands Articles d'UniversalisPas encore d'évaluation
- Conception & Modélisation CAO: Le guide ultime du débutantD'EverandConception & Modélisation CAO: Le guide ultime du débutantPas encore d'évaluation
- Semer avec succès pour rassembler avec abundance. Jardin organique et synergique: Calcul des meilleurs jours pour l'ensemencement de chaque légumeD'EverandSemer avec succès pour rassembler avec abundance. Jardin organique et synergique: Calcul des meilleurs jours pour l'ensemencement de chaque légumePas encore d'évaluation
- Harmonisation Energétique des Lieux: Habitat et haut-lieux sacrés 2020D'EverandHarmonisation Energétique des Lieux: Habitat et haut-lieux sacrés 2020Évaluation : 2.5 sur 5 étoiles2.5/5 (3)
- 20 Véritables remèdes de nos grands-mères pour maigrir vite et enfin perdre du poidsD'Everand20 Véritables remèdes de nos grands-mères pour maigrir vite et enfin perdre du poidsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Anatomie & 100 étirements essentiels: Techniques, Bénéfices attendus, Précautions à prendre, Conseils, Tableaux de séries, DouleursD'EverandAnatomie & 100 étirements essentiels: Techniques, Bénéfices attendus, Précautions à prendre, Conseils, Tableaux de séries, DouleursPas encore d'évaluation
- Le B.A.-Ba de la communication: Comment convaincre, informer, séduire ?D'EverandLe B.A.-Ba de la communication: Comment convaincre, informer, séduire ?Évaluation : 3 sur 5 étoiles3/5 (1)
- L'Ombre à l'Univers: La structure des particules élémentaires XIIfD'EverandL'Ombre à l'Univers: La structure des particules élémentaires XIIfPas encore d'évaluation
- Histoire de la psychologie scientifique: De la naissance de la psychologie à la neuropsychologie et aux champs d'application les plus actuelsD'EverandHistoire de la psychologie scientifique: De la naissance de la psychologie à la neuropsychologie et aux champs d'application les plus actuelsPas encore d'évaluation
- Géologie de l'Amérique: Les Grands Articles d'UniversalisD'EverandGéologie de l'Amérique: Les Grands Articles d'UniversalisPas encore d'évaluation
- Jus de Fruits et de Légumes Crus: 57 recettes faciles et un Guide Pratique Complet pour améliorer votre alimentation .: Santé, Vitalité et Minceur, avec ... ET DURABLEMENTD'EverandJus de Fruits et de Légumes Crus: 57 recettes faciles et un Guide Pratique Complet pour améliorer votre alimentation .: Santé, Vitalité et Minceur, avec ... ET DURABLEMENTPas encore d'évaluation
- Enseigner une Langue Etrangère Par l’Apprentissage HybrideD'EverandEnseigner une Langue Etrangère Par l’Apprentissage HybridePas encore d'évaluation
- 500 secrets pour avoir un potager merveilleuxD'Everand500 secrets pour avoir un potager merveilleuxÉvaluation : 2 sur 5 étoiles2/5 (1)
- 160 ressources pour se lancer dans la vidéo quand on n’y connait rienD'Everand160 ressources pour se lancer dans la vidéo quand on n’y connait rienPas encore d'évaluation