Vous aimerez peut-être aussi
- 05-Validation Des ProcédésDocument8 pages05-Validation Des ProcédésAbdellatif Aidoud100% (1)
- 11validation Des Procédés 2022Document5 pages11validation Des Procédés 2022Bouguetaia YacinePas encore d'évaluation
- Validation Des ProcédésDocument7 pagesValidation Des Procédésfifi fifiPas encore d'évaluation
- Validation Des ProcedesDocument12 pagesValidation Des ProcedesMorhand HizzirPas encore d'évaluation
- 309 0707 B18 Validation Des Processus Incluant Le Logiciel FRDocument7 pages309 0707 B18 Validation Des Processus Incluant Le Logiciel FRJamal MoustakimPas encore d'évaluation
- Assurance QualiteDocument65 pagesAssurance QualitefarracygmailcomPas encore d'évaluation
- BPF Chapitre 1Document10 pagesBPF Chapitre 1younessebai34Pas encore d'évaluation
- Qualification Des EquipementsDocument42 pagesQualification Des Equipementsesilab metrologiePas encore d'évaluation
- Qualification Des ÉquipementsDocument84 pagesQualification Des ÉquipementsfadliPas encore d'évaluation
- Chapitre IDocument10 pagesChapitre INotime NotimePas encore d'évaluation
- Qualification Des EquipementsDocument5 pagesQualification Des EquipementsHadjidjou Othmane100% (2)
- Système AQ PharmaceutiqueDocument31 pagesSystème AQ PharmaceutiqueAsma AmriPas encore d'évaluation
- Cours Assurance Qualité - Pour FusionDocument3 pagesCours Assurance Qualité - Pour FusionYasmine KHEDIDJAPas encore d'évaluation
- Présentation de L - Étude HACCP PDFDocument58 pagesPrésentation de L - Étude HACCP PDFAsmae ÖLkîî Jaaouane100% (1)
- Validation ProcessDocument6 pagesValidation ProcessYounes Dahmani0% (1)
- Plan Action Contrôle QualitéDocument6 pagesPlan Action Contrôle Qualitémimouni100% (1)
- 2 - Assurance Qualité Et BPFDocument7 pages2 - Assurance Qualité Et BPFmaria mariPas encore d'évaluation
- Procédés de NettoyageDocument5 pagesProcédés de Nettoyagesouheilbenazouz2Pas encore d'évaluation
- Manuel Qualite PDFDocument49 pagesManuel Qualite PDFHassan HoudoudPas encore d'évaluation
- GHTF sg3 n99 10 2004Document36 pagesGHTF sg3 n99 10 2004khaled msalbi0% (1)
- Assurance de La QualiteDocument15 pagesAssurance de La Qualitemichelboris27Pas encore d'évaluation
- Correction-TD-DUT - 2020-Abderrazzak-ELAZRIDocument6 pagesCorrection-TD-DUT - 2020-Abderrazzak-ELAZRIAbderrazzak ElazriPas encore d'évaluation
- Introduction À La Maîtrise de La Sécurité Et de La Qualité Des AlimentsDocument30 pagesIntroduction À La Maîtrise de La Sécurité Et de La Qualité Des AlimentstagadaPas encore d'évaluation
- Assurance Qualite Et Management PharmaceutiqueDocument12 pagesAssurance Qualite Et Management Pharmaceutiquesherif sherifPas encore d'évaluation
- 4 Assurance QualiteDocument52 pages4 Assurance Qualiteahmedou100% (1)
- Annexe - 15 FR DefDocument15 pagesAnnexe - 15 FR DefAmal AnanePas encore d'évaluation
- Cours BRCDocument34 pagesCours BRCamine samir100% (1)
- Iso 9001 V 2008 Cours DétailléDocument36 pagesIso 9001 V 2008 Cours DétailléHIMOUDIPas encore d'évaluation
- Aide À La ValidationDocument20 pagesAide À La ValidationkmeriemPas encore d'évaluation
- BPF ThéorieDocument38 pagesBPF ThéoriehhPas encore d'évaluation
- Management Qualité en Industrie PharmaceutiqueDocument49 pagesManagement Qualité en Industrie PharmaceutiqueWalid Masmoudi100% (1)
- Les Bonnes Pratiques de Fabrication RésuméDocument16 pagesLes Bonnes Pratiques de Fabrication RésuménourPas encore d'évaluation
- Certification QualitéDocument2 pagesCertification Qualitékarim100% (1)
- Chapitre BRCDocument21 pagesChapitre BRCasma errajiPas encore d'évaluation
- Assurance Qualité en PharmaceutiqueDocument27 pagesAssurance Qualité en PharmaceutiqueHabib TabetiPas encore d'évaluation
- Système de Management de La Qualité (SMQ)Document6 pagesSystème de Management de La Qualité (SMQ)danhahounde eudes rolland sodomePas encore d'évaluation
- Les Référentiels Qualité Et Sécurité Des Aliments Pour Les IAADocument20 pagesLes Référentiels Qualité Et Sécurité Des Aliments Pour Les IAAcedric amougouPas encore d'évaluation
- 2 - Les Systèmes de Management de La Qualité18Document12 pages2 - Les Systèmes de Management de La Qualité18Jimmy JimPas encore d'évaluation
- Explication Des QCM IPCDocument4 pagesExplication Des QCM IPCHamza ZabetPas encore d'évaluation
- BRC Global-Standard-Food Safety V8 FrenchDocument128 pagesBRC Global-Standard-Food Safety V8 Frenchdas das100% (1)
- NORMES ISO 22000 (Enregistrement Automatique)Document34 pagesNORMES ISO 22000 (Enregistrement Automatique)Dlan PulsarPas encore d'évaluation
- Support de Cours APQPDocument97 pagesSupport de Cours APQPHichem Arbi100% (3)
- Validation CCPDocument2 pagesValidation CCPnabilPas encore d'évaluation
- 1 - Contrôle Qualité Dans Les BPFDocument6 pages1 - Contrôle Qualité Dans Les BPFSou RirePas encore d'évaluation
- BPL Version 1Document71 pagesBPL Version 1Na FesPas encore d'évaluation
- SG2-03.Controle Interne QualiteDocument19 pagesSG2-03.Controle Interne QualiteZaimen sabrinaPas encore d'évaluation
- 0725 - Cours de Formation Audit ProcessDocument16 pages0725 - Cours de Formation Audit ProcessGabrielle CasanovaPas encore d'évaluation
- Système Qualité Pharmaceutique (ICH Q10) : ANSM - Mai 2013Document21 pagesSystème Qualité Pharmaceutique (ICH Q10) : ANSM - Mai 2013Mechaheb MassinissaPas encore d'évaluation
- Norme BRC Global Standard For Food Safety Issue 7 PDF FrenchDocument136 pagesNorme BRC Global Standard For Food Safety Issue 7 PDF FrenchIsmail AfifPas encore d'évaluation
- S77 Chapitre 2 Séquence 3 Référentiel BRCDocument33 pagesS77 Chapitre 2 Séquence 3 Référentiel BRCCycy HmPas encore d'évaluation
- Sujet 1Document7 pagesSujet 1Abdel BenPas encore d'évaluation
- Tests de Libération en Temps Réel Et Libération ParamétriqueDocument6 pagesTests de Libération en Temps Réel Et Libération ParamétriqueInes SahraouiPas encore d'évaluation
- Aspects Organisationnel D'un Laboratoire de Contrôle QualitéDocument34 pagesAspects Organisationnel D'un Laboratoire de Contrôle QualitésalimPas encore d'évaluation
- AQ - IN01. Ed 2 Elaboration D'un Plan de SurveillanceDocument10 pagesAQ - IN01. Ed 2 Elaboration D'un Plan de Surveillancemohamed bchihyPas encore d'évaluation
- Fiche de PosteDocument4 pagesFiche de PostekhaledPas encore d'évaluation
- La Norme ISO 22000 Sécurité Des Denrées AlimentairesDocument37 pagesLa Norme ISO 22000 Sécurité Des Denrées AlimentairesYassine Alazizi100% (3)
- La Méthode SCORE: Mesurer et améliorer les performances opérationnellesD'EverandLa Méthode SCORE: Mesurer et améliorer les performances opérationnellesPas encore d'évaluation
- Les inducteurs de l'amélioration continue: Plus de 365 pistes de progrès au service de la performance durable de votre entrepriseD'EverandLes inducteurs de l'amélioration continue: Plus de 365 pistes de progrès au service de la performance durable de votre entreprisePas encore d'évaluation
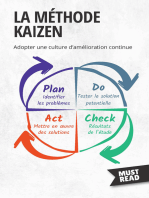
- La Méthode Kaizen: Adopter une culture d'amélioration continueD'EverandLa Méthode Kaizen: Adopter une culture d'amélioration continuePas encore d'évaluation
- La théorie des contraintes: Identifier et éliminer les obstacles pour une efficacité maximaleD'EverandLa théorie des contraintes: Identifier et éliminer les obstacles pour une efficacité maximalePas encore d'évaluation
- Retinopathie HypertensiveDocument28 pagesRetinopathie HypertensiveMelissa MenasriaPas encore d'évaluation
- Galenique 3eme EMDDocument7 pagesGalenique 3eme EMDMelissa MenasriaPas encore d'évaluation
- Les QuinquinasDocument31 pagesLes QuinquinasMelissa MenasriaPas encore d'évaluation
- Orcinols Et PhloroglucinolsDocument49 pagesOrcinols Et PhloroglucinolsMelissa MenasriaPas encore d'évaluation
- ANATOMIE ET PHYSIOLOGIE DE LA GLANDE MAMMAIRE ParamedicalDocument37 pagesANATOMIE ET PHYSIOLOGIE DE LA GLANDE MAMMAIRE ParamedicalMelissa MenasriaPas encore d'évaluation
- Alcaloïde IndoloisopréniqueDocument33 pagesAlcaloïde IndoloisopréniqueMelissa MenasriaPas encore d'évaluation
- Galenique 3eme EMDDocument7 pagesGalenique 3eme EMDMelissa MenasriaPas encore d'évaluation
- Alcaloïdes Indolomonoterpenique Pervenche Et RawolfiasDocument39 pagesAlcaloïdes Indolomonoterpenique Pervenche Et RawolfiasMelissa MenasriaPas encore d'évaluation
- 12 - Recevabilité D'une OrdannanceDocument6 pages12 - Recevabilité D'une OrdannanceMelissa MenasriaPas encore d'évaluation
- Methodes D'extraction Et D'analyse ToxicologiqueDocument9 pagesMethodes D'extraction Et D'analyse ToxicologiqueMelissa MenasriaPas encore d'évaluation
- 05 - Encadrement de La Profession PharmaceutiqueDocument7 pages05 - Encadrement de La Profession PharmaceutiqueMelissa MenasriaPas encore d'évaluation
- 13 - Ethers de GlycolDocument39 pages13 - Ethers de GlycolMelissa MenasriaPas encore d'évaluation
- Toxicologie Professionnelle Et Environnementale (C21)Document1 pageToxicologie Professionnelle Et Environnementale (C21)Melissa MenasriaPas encore d'évaluation
- 14 - Solvants ChlorésDocument24 pages14 - Solvants ChlorésMelissa MenasriaPas encore d'évaluation
- Ecg - TabaryDocument11 pagesEcg - TabaryLucy RosewoodPas encore d'évaluation
- 14-Généralités Sur Les Antibiotiques Et Principes D'utilisation - PR K.reggABIDocument35 pages14-Généralités Sur Les Antibiotiques Et Principes D'utilisation - PR K.reggABIAlex JuvePas encore d'évaluation
- Bilan TVT 2019Document14 pagesBilan TVT 2019Fred AliPas encore d'évaluation
- Memoire Malla Talla 2 28112023Document70 pagesMemoire Malla Talla 2 28112023Stephane KiagePas encore d'évaluation
- ATHLETISME Sauts Et Lancers À Sens Le 19 Juin 2021Document3 pagesATHLETISME Sauts Et Lancers À Sens Le 19 Juin 2021HUARDPas encore d'évaluation
- Lois Sur Le Climat Des Affaires en RDCDocument414 pagesLois Sur Le Climat Des Affaires en RDCguymbulaPas encore d'évaluation
- CardDocument2 pagesCardclement JullienPas encore d'évaluation
- Soins Infirmiers Dans Les Maladies Infectieuses Et Parasitaires - CopierDocument10 pagesSoins Infirmiers Dans Les Maladies Infectieuses Et Parasitaires - CopierOsman Aden89% (9)
- Cas Clinique 1Document15 pagesCas Clinique 1iras2010100% (1)
- Semaine Africaine N°4056Document16 pagesSemaine Africaine N°4056Rudolph OzalaPas encore d'évaluation
- FR FR 117349e Aniosyme DLM Maxi (117349e)Document15 pagesFR FR 117349e Aniosyme DLM Maxi (117349e)Jean GomisPas encore d'évaluation
- SAP-FTO-38-Relevage D'une Victime en Position ParticulièreDocument4 pagesSAP-FTO-38-Relevage D'une Victime en Position Particulièreomar benounaPas encore d'évaluation
- Les Ostéopathie1Document5 pagesLes Ostéopathie1Walid KouidriPas encore d'évaluation
- Ast Got 4 1 SL ElitechgroupDocument4 pagesAst Got 4 1 SL ElitechgroupAnonymous lK8PC1IrlOPas encore d'évaluation
- Liste Des Prix Convention Avec Le Laboratoire D'analyses Médicales MODERNELAMDocument8 pagesListe Des Prix Convention Avec Le Laboratoire D'analyses Médicales MODERNELAMbilel oukilPas encore d'évaluation
- 31-Souffrance Få Tale AigueDocument54 pages31-Souffrance Få Tale Aiguezianiikram0673Pas encore d'évaluation
- TD - PauvretéDocument7 pagesTD - PauvretéturroPas encore d'évaluation
- Cancer de La ProstateDocument4 pagesCancer de La ProstateMohamed KadiPas encore d'évaluation
- Autres Questions PoséesDocument1 pageAutres Questions PoséesLou AizePas encore d'évaluation
- Bilan Internat.1Document7 pagesBilan Internat.1Samira BoulesnanePas encore d'évaluation
- Guide Des Evenements Solidaires AFSA 2017-1497881153Document76 pagesGuide Des Evenements Solidaires AFSA 2017-1497881153Jacphi NoukpoPas encore d'évaluation
- 2 - Étude Par Questionnaire de La Connaissance Des Facteurs de Risques Psychosociaux Et de L'exercice en Accès Direct Pour La Lombalgie Commune Chez Les Masseurs-Kinésithérapeutes LibérauxDocument8 pages2 - Étude Par Questionnaire de La Connaissance Des Facteurs de Risques Psychosociaux Et de L'exercice en Accès Direct Pour La Lombalgie Commune Chez Les Masseurs-Kinésithérapeutes LibérauxYANETH PATRICIA BARÓN OSORIOPas encore d'évaluation