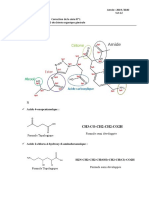
Vous aimerez peut-être aussi
- Cours Réactivité Chimique - C122Document75 pagesCours Réactivité Chimique - C122DARIF AYMANPas encore d'évaluation
- 1.2.1-Exercices OsesDocument1 page1.2.1-Exercices OsesLy Dia100% (1)
- BioelectricitéDocument9 pagesBioelectricitéMedMan6100% (1)
- Chimie ch8Document10 pagesChimie ch8nadpharm13Pas encore d'évaluation
- TD1 AtomistiqueDocument4 pagesTD1 AtomistiquerochdibaatiPas encore d'évaluation
- DM en Plein Coeur Enoncé-CorrigéDocument8 pagesDM en Plein Coeur Enoncé-CorrigédalyyyamaraPas encore d'évaluation
- Biochimie-Ue1: PacesDocument23 pagesBiochimie-Ue1: Pacesludovic pacmogdaPas encore d'évaluation
- TD-chimie OrgaDocument21 pagesTD-chimie OrgaJean AnanPas encore d'évaluation
- Paul Arnaud 19a PDFDocument3 pagesPaul Arnaud 19a PDFBoubs SelkaPas encore d'évaluation
- Réticulum Endoplasmique OranDocument7 pagesRéticulum Endoplasmique OranHaitam El OuahabiPas encore d'évaluation
- Mecanismes Reactionnels PDFDocument6 pagesMecanismes Reactionnels PDFJérémie Nimpagaritse100% (1)
- Emd 1 de Chimie Première Année Médecine Et Médecine DentaireDocument3 pagesEmd 1 de Chimie Première Année Médecine Et Médecine Dentaireσεφ ΙσλάμPas encore d'évaluation
- Equilibre Acido BasiqueDocument22 pagesEquilibre Acido Basiquewarda oudghiriPas encore d'évaluation
- Les Epitheliums GlandulairesDocument112 pagesLes Epitheliums GlandulairesAbdoulaziz SidilaminePas encore d'évaluation
- Chimie Générale - Pcem1Document128 pagesChimie Générale - Pcem1Lo RanPas encore d'évaluation
- Modélisation Moléculaire Cours 2021Document120 pagesModélisation Moléculaire Cours 2021CHEIKH ABDOUL AZIZ H'MEIDYPas encore d'évaluation
- NomenclatureDocument10 pagesNomenclatureKallel JihenePas encore d'évaluation
- Solvants en Chimie OrganiqueDocument3 pagesSolvants en Chimie OrganiqueMvone100% (2)
- 12 PeroxysomesDocument5 pages12 PeroxysomesImen Mestar100% (1)
- Serie de TD ChimieDocument5 pagesSerie de TD Chimieraouf teach 04Pas encore d'évaluation
- Poly Ziko Et HibaDocument89 pagesPoly Ziko Et HibaSalwa CatPas encore d'évaluation
- Resume Complet La Thermodynamique PDFDocument11 pagesResume Complet La Thermodynamique PDFAL HaniPas encore d'évaluation
- Série N°1-CorrigéeDocument9 pagesSérie N°1-CorrigéeZineb DahmaniPas encore d'évaluation
- Chimie SolutionDocument164 pagesChimie SolutionZonta NeoPas encore d'évaluation
- Acidite D Un VinaigreDocument6 pagesAcidite D Un Vinaigreboudriat ahmedPas encore d'évaluation
- Biophysique Des Solutions 2023Document26 pagesBiophysique Des Solutions 2023zaki taleb100% (1)
- UE31 BPH03 Boi Memb 2Document65 pagesUE31 BPH03 Boi Memb 2Christian DuboisPas encore d'évaluation
- TD Nomenclature Corrigé12goodprepa PDFDocument3 pagesTD Nomenclature Corrigé12goodprepa PDFNour EddinePas encore d'évaluation
- Biophysique Cours 1Document105 pagesBiophysique Cours 1Smail FreeManPas encore d'évaluation
- TD 4 Biophysique CorrigDocument5 pagesTD 4 Biophysique CorrigWiwi LauraPas encore d'évaluation
- Correction Des Travaux Dirigés: Exercice N°1Document2 pagesCorrection Des Travaux Dirigés: Exercice N°1Abdullah MohamedPas encore d'évaluation
- PCEM1 Physio Renale Objectifs Plan Dec Ours RudimentsDocument8 pagesPCEM1 Physio Renale Objectifs Plan Dec Ours RudimentsGhassen SouissiPas encore d'évaluation
- PC 1ère CD-C2 Hydrocarbures SaturésDocument11 pagesPC 1ère CD-C2 Hydrocarbures SaturésTouré Kiyofo HabibPas encore d'évaluation
- Potentiel ChimiqueDocument1 pagePotentiel ChimiqueJob ElsonPas encore d'évaluation
- 9-Système Renine Angiotensine AldostéroneDocument30 pages9-Système Renine Angiotensine AldostéroneRaïssa sawadogoPas encore d'évaluation
- TD RespirationDocument7 pagesTD RespirationBushaPas encore d'évaluation
- Application de La Pression Osmotique PDFDocument4 pagesApplication de La Pression Osmotique PDFZied BouabenePas encore d'évaluation
- 2-Le Tissu ConjonctifDocument92 pages2-Le Tissu ConjonctifYouness Khalfaoui100% (3)
- Série 2 Oxydo RéductionDocument8 pagesSérie 2 Oxydo RéductionSection E GPPas encore d'évaluation
- Biophysique TD Corriges Diffusion Dans La Phase LiquidesDocument4 pagesBiophysique TD Corriges Diffusion Dans La Phase LiquidesYacine AchPas encore d'évaluation
- Equilibre Acide-Base Et Ses TroublesDocument25 pagesEquilibre Acide-Base Et Ses TroublesSOLTANIPas encore d'évaluation
- Examen 20141 SDocument7 pagesExamen 20141 SElbachaPas encore d'évaluation
- Stereochimie Et Reactivite en Serie CycliqueDocument9 pagesStereochimie Et Reactivite en Serie CycliqueJean-François Abena100% (1)
- Cytogenetique pptx-1Document88 pagesCytogenetique pptx-1franck brice badoPas encore d'évaluation
- Tableau D'anatomie ComparéeDocument3 pagesTableau D'anatomie Comparéehalack100% (1)
- Nomeclature Des Composés OrganiquesDocument32 pagesNomeclature Des Composés OrganiquesmehdiPas encore d'évaluation
- UploadedFile 131951726939398212Document53 pagesUploadedFile 131951726939398212AhmedPas encore d'évaluation
- Exercices Lois Discrètes Usuelle S CorrigéDocument5 pagesExercices Lois Discrètes Usuelle S CorrigéTazouta YassinePas encore d'évaluation
- Cours Lipides - Part 1Document74 pagesCours Lipides - Part 1AhmedPas encore d'évaluation
- Essentiel-Conditionnement Du MedicamentDocument2 pagesEssentiel-Conditionnement Du MedicamentMustapha Mektan100% (1)
- Polycopié de Biophysique-28!03!2020Document77 pagesPolycopié de Biophysique-28!03!2020Oumia HarbitPas encore d'évaluation
- 2 AlcenesDocument8 pages2 Alcenesam47Pas encore d'évaluation
- Exercices AtomistiqueDocument4 pagesExercices AtomistiqueZakaria AlbertoPas encore d'évaluation
- Rapport Acide BaseDocument10 pagesRapport Acide BaseBouafia AbdelrahmanePas encore d'évaluation
- CF Chimie Des Solutions SMPC2 Ratrappage 2016Document4 pagesCF Chimie Des Solutions SMPC2 Ratrappage 2016Hamza m'rabet100% (2)
- CF Chimie Des Solutions SMPC2 Ratrappage 2016Document4 pagesCF Chimie Des Solutions SMPC2 Ratrappage 2016Nora AronPas encore d'évaluation
- Abdelali AhDocument33 pagesAbdelali Ahcerveaumental80Pas encore d'évaluation
- Marolet MaDocument5 pagesMarolet MamedsalemeddahPas encore d'évaluation
- Chapitre IIDocument12 pagesChapitre IIPascal GadedjissoPas encore d'évaluation
- Health and Safety Heat Stress Fact Sheet-V3-FrDocument13 pagesHealth and Safety Heat Stress Fact Sheet-V3-Frmagloire amivaPas encore d'évaluation
- Master BinziDocument102 pagesMaster Binzimagloire amivaPas encore d'évaluation
- Iso 999 1996Document13 pagesIso 999 1996magloire amivaPas encore d'évaluation
- Rmstpub 25 32 PP Dtermination Acide Cyanhydrique ManiocDocument8 pagesRmstpub 25 32 PP Dtermination Acide Cyanhydrique Maniocmagloire amivaPas encore d'évaluation
- RR 39126 FRDocument46 pagesRR 39126 FRmagloire amivaPas encore d'évaluation
- Fiche Sur La Cassava: Programme "Desira - Development Smart Innovation Through Research inDocument19 pagesFiche Sur La Cassava: Programme "Desira - Development Smart Innovation Through Research inmagloire amivaPas encore d'évaluation
- Pdfcours 4. PhtamponsDocument3 pagesPdfcours 4. Phtamponsmagloire amivaPas encore d'évaluation
- Pilard Etienne 2021Document245 pagesPilard Etienne 2021magloire amivaPas encore d'évaluation
- GardeDocument8 pagesGardemagloire amivaPas encore d'évaluation
- TP 1: Réaction D'oxydo-Réduction: Couple Oxydant / RéducteurDocument2 pagesTP 1: Réaction D'oxydo-Réduction: Couple Oxydant / Réducteurmagloire amivaPas encore d'évaluation
- TP4 DosageparoxydorductionDocument12 pagesTP4 Dosageparoxydorductionhamranibaha44Pas encore d'évaluation
- andrianarisonIreneM SN M2 12Document112 pagesandrianarisonIreneM SN M2 12magloire amivaPas encore d'évaluation
- L Evaluation Des Projets D Eau Potable EDocument9 pagesL Evaluation Des Projets D Eau Potable Emagloire amivaPas encore d'évaluation
- Adt Bap B Chimie 2004 Epreuve Pro 1172585383064Document4 pagesAdt Bap B Chimie 2004 Epreuve Pro 1172585383064magloire amivaPas encore d'évaluation
- 0.81.03.89 Convention Du 22 Mars 1989 Dechets Dangereux Controle Des MouvementsDocument60 pages0.81.03.89 Convention Du 22 Mars 1989 Dechets Dangereux Controle Des Mouvementsmagloire amivaPas encore d'évaluation
- Struct Mat DZ PDFDocument86 pagesStruct Mat DZ PDFMokhtarBensaidPas encore d'évaluation
- 2.9.23. Masse Volumique DesDocument2 pages2.9.23. Masse Volumique Desmagloire amivaPas encore d'évaluation
- P 52 À 61 TP Acidité Volatile Du Vin BTSA 1 ANABIOTECDocument10 pagesP 52 À 61 TP Acidité Volatile Du Vin BTSA 1 ANABIOTECmagloire amivaPas encore d'évaluation
- NAT 1reSTL D2VinaigreDocument10 pagesNAT 1reSTL D2Vinaigremagloire amivaPas encore d'évaluation
- L'analyse Du Vin: A - Détermination Du Degré Alcoolique Du VinDocument15 pagesL'analyse Du Vin: A - Détermination Du Degré Alcoolique Du Vinmagloire amivaPas encore d'évaluation
- M1 S1UE1 - VALLARD - Gestion Déchets 22-23Document3 pagesM1 S1UE1 - VALLARD - Gestion Déchets 22-23magloire amivaPas encore d'évaluation
- Hardness of WaterDocument5 pagesHardness of Watermagloire amivaPas encore d'évaluation
- 2.9.23. Masse Volumique DesDocument2 pages2.9.23. Masse Volumique Desmagloire amivaPas encore d'évaluation
- Repetition Premiere Masse MolaireDocument1 pageRepetition Premiere Masse Molairemagloire amivaPas encore d'évaluation
- TP Ndeg3semestre 1Document3 pagesTP Ndeg3semestre 1magloire amivaPas encore d'évaluation
- Liste Des Candidats Admis A Lecrit Concours Numerique 2023 DIR CDDocument2 pagesListe Des Candidats Admis A Lecrit Concours Numerique 2023 DIR CDmagloire amivaPas encore d'évaluation
- Fitta - GheDeir-AmarDocument56 pagesFitta - GheDeir-Amarmagloire amivaPas encore d'évaluation
- Etat de Besoin de Chimie Générale Prépa ST - 103629Document1 pageEtat de Besoin de Chimie Générale Prépa ST - 103629magloire amivaPas encore d'évaluation
- CATALOGUE DES MATIERES PREMIERES 2-Fi15997396 PDFDocument44 pagesCATALOGUE DES MATIERES PREMIERES 2-Fi15997396 PDFmagloire amivaPas encore d'évaluation
- Liste Des Candidats Admis A Lecrit Concours Numerique 2023 ATA 2 PDFDocument61 pagesListe Des Candidats Admis A Lecrit Concours Numerique 2023 ATA 2 PDFmagloire amivaPas encore d'évaluation
- Cours de Chimie Organique AAT Filière Bois 2022-2023Document36 pagesCours de Chimie Organique AAT Filière Bois 2022-2023Bella BellaPas encore d'évaluation
- TP Chimie Organique Synthèse Du 7,7-DichloronorcaraneDocument6 pagesTP Chimie Organique Synthèse Du 7,7-DichloronorcaraneEm's NonoPas encore d'évaluation
- Bac S 2015 Amerique Nord Ex 2 PDFDocument5 pagesBac S 2015 Amerique Nord Ex 2 PDFderic fekekangPas encore d'évaluation
- ChapSA 4 PrecipitationDocument22 pagesChapSA 4 PrecipitationOumar CamaraPas encore d'évaluation
- 2005 Maroc Spe CorrectionDocument2 pages2005 Maroc Spe CorrectionNada LaaballiPas encore d'évaluation
- Preparation Des Candidats A L'Examen Du Bac 2023: Elements de Reponses ADocument7 pagesPreparation Des Candidats A L'Examen Du Bac 2023: Elements de Reponses AAgasroPas encore d'évaluation
- Serie 9 Acides Alpha - AminesDocument2 pagesSerie 9 Acides Alpha - Amineskathy100% (1)
- Dosage Azote SolDocument22 pagesDosage Azote Solkamal100% (1)
- Chapitre II Acides Et Bases 2021-2022 Partie 1Document11 pagesChapitre II Acides Et Bases 2021-2022 Partie 1KHEDIM MouradPas encore d'évaluation
- Synthèse Du BENZHYDROL DiphènylmèthanolDocument6 pagesSynthèse Du BENZHYDROL DiphènylmèthanolHoussem Eddine KAFI100% (1)
- Oxydoréduction PC, Énoncés Des ExercicesDocument30 pagesOxydoréduction PC, Énoncés Des ExercicesLili KhePas encore d'évaluation
- Actions Acides Sur Plomb Et CuivreDocument5 pagesActions Acides Sur Plomb Et CuivreBiranePas encore d'évaluation
- Cours de Chimie en Solution Les Acides Et Les Bases: 1 Année Biologie 2019-2020Document37 pagesCours de Chimie en Solution Les Acides Et Les Bases: 1 Année Biologie 2019-2020kheddirachidPas encore d'évaluation
- Manuel de Phototypie Par M. G. BONNET - 1889Document155 pagesManuel de Phototypie Par M. G. BONNET - 1889Michel MOMAL100% (1)
- Oxydoréduction PDFDocument5 pagesOxydoréduction PDFGaniyou AdenidjiPas encore d'évaluation
- Resines PDFDocument14 pagesResines PDFboutheina benncirPas encore d'évaluation
- P 52 À 61 TP Acidité Volatile Du Vin BTSA 1 ANABIOTECDocument10 pagesP 52 À 61 TP Acidité Volatile Du Vin BTSA 1 ANABIOTECmagloire amivaPas encore d'évaluation
- Les Analyses Physico Chimiques Des Eaux NaturellesDocument21 pagesLes Analyses Physico Chimiques Des Eaux NaturellesDahou AliPas encore d'évaluation
- Acide ChlorhydriqueDocument22 pagesAcide ChlorhydriqueFrederic WustPas encore d'évaluation
- 14 - Solvants ChlorésDocument24 pages14 - Solvants ChlorésMelissa MenasriaPas encore d'évaluation
- Couple Acide - Base: ExercicesDocument16 pagesCouple Acide - Base: ExercicesAhamadi ElhouyounPas encore d'évaluation
- Stereochimie Et Reactivite en Serie CycliqueDocument9 pagesStereochimie Et Reactivite en Serie CycliqueJean-François Abena100% (1)
- UE1-Biochimie-12-Métabo AAsDocument17 pagesUE1-Biochimie-12-Métabo AAssun-nee-chan9Pas encore d'évaluation
- Crème Hydratante Visage Bio À L'Acide HyaluroniquDocument2 pagesCrème Hydratante Visage Bio À L'Acide HyaluroniquCraxeenePas encore d'évaluation
- Anyalyses Du MielsDocument16 pagesAnyalyses Du MielsHanin CarolePas encore d'évaluation
- T.P 01 de Chimie Des EauxDocument10 pagesT.P 01 de Chimie Des EauxBòû ChŔâ Ñe50% (2)
- Oxydation Et RéductionDocument2 pagesOxydation Et RéductionFélix Kouassi100% (1)
- EstérificationDocument14 pagesEstérificationFrederic WustPas encore d'évaluation
- Chapitre 2 - Equilibrage Des Reactions RedoxDocument2 pagesChapitre 2 - Equilibrage Des Reactions RedoxJoe FinianosPas encore d'évaluation
- BIOCHIMIEDocument5 pagesBIOCHIMIEMamoutou BouaréPas encore d'évaluation