Vous aimerez peut-être aussi
- Commission du Codex Alimentarius: Manuel de Procédure Vingt-sixième editionD'EverandCommission du Codex Alimentarius: Manuel de Procédure Vingt-sixième editionÉvaluation : 5 sur 5 étoiles5/5 (1)
- Exigences de La Norme Iso 22000 Version 2018Document35 pagesExigences de La Norme Iso 22000 Version 2018ARKASPas encore d'évaluation
- Intégrer le genre dans la mise en oeuvre et le suivi-évaluation des programmes de transferts en espèces et de travaux publics: Guide technique 3 de la FAOD'EverandIntégrer le genre dans la mise en oeuvre et le suivi-évaluation des programmes de transferts en espèces et de travaux publics: Guide technique 3 de la FAOPas encore d'évaluation
- Mise en Place Du Système HACCP Pour La Ligne de Production de La Viande Hachée Cuite Au Sein Du Restaurant Universitaire Sais 1Document48 pagesMise en Place Du Système HACCP Pour La Ligne de Production de La Viande Hachée Cuite Au Sein Du Restaurant Universitaire Sais 1Hajar OuPas encore d'évaluation
- Procedure Retrait Rappel - V2020Document3 pagesProcedure Retrait Rappel - V2020casperPas encore d'évaluation
- AFNOR-ISO22000-Module-n9-Validation Des Mesures de Maitrise Et de ProcedeDocument10 pagesAFNOR-ISO22000-Module-n9-Validation Des Mesures de Maitrise Et de Procedehana100% (3)
- Plan de ControDocument4 pagesPlan de ControabdellahPas encore d'évaluation
- Fiche Action Programme Exigences de La Norme FSSC 22000 PDFDocument2 pagesFiche Action Programme Exigences de La Norme FSSC 22000 PDFSidouPas encore d'évaluation
- EXIGENCES ISO 13485 Version 2016Document37 pagesEXIGENCES ISO 13485 Version 2016najwa zinaoui100% (1)
- Non ConformitéDocument11 pagesNon Conformitéhiba ben saih100% (1)
- 0PR015A1 Audit Produit en ProductionDocument5 pages0PR015A1 Audit Produit en ProductionNizar EnnettaPas encore d'évaluation
- Examen 17025 - RattrapageDocument5 pagesExamen 17025 - RattrapageMery ZehPas encore d'évaluation
- Contribution À La Mise en Place Du Système de Management de La Sécurité Des Denrées Alimentaires Suivant La Norme ISO 22000 - V20Document59 pagesContribution À La Mise en Place Du Système de Management de La Sécurité Des Denrées Alimentaires Suivant La Norme ISO 22000 - V20Kamal WadihPas encore d'évaluation
- Sap-Pr-002 Procedure de Gest° Des Emballages Non ConformesDocument2 pagesSap-Pr-002 Procedure de Gest° Des Emballages Non ConformesGondwanais LamdaPas encore d'évaluation
- Manuel Qualité ZEURGDocument34 pagesManuel Qualité ZEURGالحمد لله100% (1)
- Grille Dvaluation Systme Qualit ISO 9001 2015Document23 pagesGrille Dvaluation Systme Qualit ISO 9001 2015KalianaPas encore d'évaluation
- Check Liste HACCP Mensuelle Vétérinaire de ControlDocument2 pagesCheck Liste HACCP Mensuelle Vétérinaire de Controlel biache100% (1)
- Procedure Action CorrectiveDocument5 pagesProcedure Action CorrectiveEL Merniss AzizaPas encore d'évaluation
- Module Soutien Iso22000 10 Tracabilite Et Gestion de CriseDocument11 pagesModule Soutien Iso22000 10 Tracabilite Et Gestion de CriseToufik DehilisPas encore d'évaluation
- Initiation À La Mise en Place de La Norme ISO 22000 Version 2018 Au Sein de S.A.R.L. GERBIOR Groupe Benhamadi Bordj Bou A - 1Document52 pagesInitiation À La Mise en Place de La Norme ISO 22000 Version 2018 Au Sein de S.A.R.L. GERBIOR Groupe Benhamadi Bordj Bou A - 1Zouhour Hannachi100% (2)
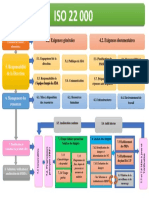
- Carte Conceptuelle ISO 22000Document1 pageCarte Conceptuelle ISO 22000LabroufiAbdelouahed33% (3)
- Exemple de Plan HACCP Pour Un RestaurantDocument107 pagesExemple de Plan HACCP Pour Un RestaurantScribdTranslationsPas encore d'évaluation
- Rapport SWOT Durthel FichierDocument19 pagesRapport SWOT Durthel FichierDurthel100% (1)
- Contribution À La Mise en Place de La Démarche HACCP Pour La Levure Sèche À LESAFFRE-Maroc - Ghita BENMOUSSADocument36 pagesContribution À La Mise en Place de La Démarche HACCP Pour La Levure Sèche À LESAFFRE-Maroc - Ghita BENMOUSSAyassine DAOUDPas encore d'évaluation
- Eexigence ISO 22000Document35 pagesEexigence ISO 22000Yahya AlferdaoussPas encore d'évaluation
- FSSC-22000-Francais Version6Document83 pagesFSSC-22000-Francais Version6honorabossladyPas encore d'évaluation
- Procédure "GESTION DES DECHETS"Document6 pagesProcédure "GESTION DES DECHETS"Soufian ErramizyPas encore d'évaluation
- NM 08 0 002 (Haccp)Document15 pagesNM 08 0 002 (Haccp)elalaoui25100% (2)
- Exigences de La Norme Iso 22000 Version 2018Document34 pagesExigences de La Norme Iso 22000 Version 2018gratracrumadde-6168Pas encore d'évaluation
- Manuel QualitéDocument12 pagesManuel QualitémguissePas encore d'évaluation
- État Avancement IATFDocument5 pagesÉtat Avancement IATFNihel MerhbenPas encore d'évaluation
- Formation HalalDocument83 pagesFormation HalalNajib tounsiPas encore d'évaluation
- Contribution À La Mise en Place Du Système de Management de La Sécurité Des Denrées Alimentaires Suivant La Norme ISO 22000 - V2Document57 pagesContribution À La Mise en Place Du Système de Management de La Sécurité Des Denrées Alimentaires Suivant La Norme ISO 22000 - V2yassinelakhnati344Pas encore d'évaluation
- Fiche Des OpérateursDocument2 pagesFiche Des OpérateursProdiak QualityPas encore d'évaluation
- Chek-List Audit FSSC 22000 V 5.1Document27 pagesChek-List Audit FSSC 22000 V 5.1NiassPas encore d'évaluation
- Preparation A La Mise en Place - Ikrame RIBAT - 4276Document55 pagesPreparation A La Mise en Place - Ikrame RIBAT - 4276ARKASPas encore d'évaluation
- Article Maîtrise Des Modifications Dans La Norme ISO 9001:2015Document4 pagesArticle Maîtrise Des Modifications Dans La Norme ISO 9001:2015Abderrahman Oueslati100% (1)
- Tableau 6.3 Liste Des Enregistrements Cités Dans l'ISO 22000 Enregistrements RéférenceDocument92 pagesTableau 6.3 Liste Des Enregistrements Cités Dans l'ISO 22000 Enregistrements RéférencehhPas encore d'évaluation
- Procedure Non Conformite Actionp AP AcDocument6 pagesProcedure Non Conformite Actionp AP AcAkimBi100% (1)
- Analyse Des Dangers 2020Document50 pagesAnalyse Des Dangers 2020BEN ABBES100% (1)
- Procedure Controle Reception Et Gestion Des Non Conformites FournisseursDocument8 pagesProcedure Controle Reception Et Gestion Des Non Conformites FournisseursAbderrahmane79Pas encore d'évaluation
- Iso 22000Document21 pagesIso 22000Nour MassoudiPas encore d'évaluation
- Memoire Raw ActsDocument75 pagesMemoire Raw ActsApolion DjoundaPas encore d'évaluation
- PR-Manuel QualitéDocument11 pagesPR-Manuel QualitéSoulaimaPas encore d'évaluation
- Liste de Diffusion Des Documents Du SMéDocument2 pagesListe de Diffusion Des Documents Du SMéAmine RamziPas encore d'évaluation
- Audit BPF Labo CQDocument2 pagesAudit BPF Labo CQFaress RabiPas encore d'évaluation
- ISO 22000 Module de Soutien N 5 La DocumentationDocument6 pagesISO 22000 Module de Soutien N 5 La DocumentationRedouan Lerhrissi50% (2)
- Prpo PDFDocument89 pagesPrpo PDFLA HadjerPas encore d'évaluation
- StagaireDocument79 pagesStagaireawatef0% (1)
- Rapport Haccp VFDocument60 pagesRapport Haccp VFZineb GoubarPas encore d'évaluation
- Eexigence ISO 22000Document34 pagesEexigence ISO 22000Marsit Med AminePas encore d'évaluation
- Guide HaccpDocument0 pageGuide HaccpJaykishan ParmarPas encore d'évaluation
- La Procédure de Maîtrise Documentaire Répondant Aux Exigences ISODocument9 pagesLa Procédure de Maîtrise Documentaire Répondant Aux Exigences ISOماهر زارعي0% (1)
- Modele Manuel PRPDocument22 pagesModele Manuel PRPjanati100% (1)
- P001 - Maitrise Des Docs Et EnregistrementsDocument6 pagesP001 - Maitrise Des Docs Et EnregistrementsColombe CérèsPas encore d'évaluation
- Manuel Qualité AgroalimentaireDocument4 pagesManuel Qualité AgroalimentaireAgroAliment.net100% (1)
- Iso22716 AutodiagnosticDocument17 pagesIso22716 AutodiagnosticMedali BouazizPas encore d'évaluation
- Comparaison 22000 Et ISO 9001Document20 pagesComparaison 22000 Et ISO 9001Oumaima Trabelsi100% (2)
- FSSC 22000Document75 pagesFSSC 22000najwa zinaouiPas encore d'évaluation
- PUB100377 FRDocument12 pagesPUB100377 FRSoraya Bouakkaz100% (1)
- 406 - SysML en Action Avec Cameo Systems Modeler - Casse 2 - Première PartieDocument11 pages406 - SysML en Action Avec Cameo Systems Modeler - Casse 2 - Première PartieSalahdinePas encore d'évaluation
- Chapitre 02 2022 - 2023Document46 pagesChapitre 02 2022 - 2023Thï ZïrīPas encore d'évaluation
- Referentiel CFR BABOKv3 V1.0Document15 pagesReferentiel CFR BABOKv3 V1.0Badri Radhi0% (1)
- French 20171001 IATF 16949 FAQ - Octobre 2017Document11 pagesFrench 20171001 IATF 16949 FAQ - Octobre 2017aminos85Pas encore d'évaluation
- Cours de Gnie Logiciel-Boudaa BoudjemaaDocument215 pagesCours de Gnie Logiciel-Boudaa BoudjemaaMarcelin KamseuPas encore d'évaluation
- IFS Food Defense Guidelines - FR - 2012 - 02 - 21Document15 pagesIFS Food Defense Guidelines - FR - 2012 - 02 - 21nicolas100% (1)
- Cigi09 120Document8 pagesCigi09 120Rihab SadikiPas encore d'évaluation
- Rapport de Projet de Fin D'études: TitreDocument75 pagesRapport de Projet de Fin D'études: TitreSAWYERPas encore d'évaluation
- Parcours C QT Sii LearningDocument15 pagesParcours C QT Sii LearningÀymén ChebbiPas encore d'évaluation
- Analyse Besoins Développement Logiciel: Des Pour LeDocument26 pagesAnalyse Besoins Développement Logiciel: Des Pour LeKITSONPas encore d'évaluation
- Qualité Automobile Les Engagements Qualité Et Fiabilité de PSADocument8 pagesQualité Automobile Les Engagements Qualité Et Fiabilité de PSAOumaima BenhamedPas encore d'évaluation
- Fermetures: Commerciales & IndustriellesDocument63 pagesFermetures: Commerciales & Industrielleshrms YepezPas encore d'évaluation
- Iso 9000Document18 pagesIso 9000Hamdi NefetiPas encore d'évaluation
- PAQ - Plan Assurance Qualité - v1.0 - 20110515Document44 pagesPAQ - Plan Assurance Qualité - v1.0 - 20110515elamigosolitario100% (3)
- MMOGLE v4 FrancaisDocument23 pagesMMOGLE v4 FrancaisAdriana Hublea100% (1)
- FSSC V5 LA TrainingDocument43 pagesFSSC V5 LA Trainingsajid waqas100% (2)
- Diapo Stagiaire SysmlDocument17 pagesDiapo Stagiaire SysmlPhilippe RivesPas encore d'évaluation
- Cours 2 Travailler en ÉquipeDocument33 pagesCours 2 Travailler en ÉquipeWafa FradiPas encore d'évaluation
- Rapport de Stage MarieDocument27 pagesRapport de Stage MarieChikara Gabriel Edou100% (1)
- 4 - Specification Du SystemeDocument54 pages4 - Specification Du SystemeOuiamePas encore d'évaluation
- MaintenanceDocument17 pagesMaintenancemanofdamisi25 DamisiPas encore d'évaluation
- 10-Fiche 9 444 445 453 Documentation Enregistrements V2Document13 pages10-Fiche 9 444 445 453 Documentation Enregistrements V2adam_3000Pas encore d'évaluation
- Une Politique de Gestion Des Documents D'activité Pour Une Gouvernance Documentaire Stratégique Documentaire StrategiqueDocument20 pagesUne Politique de Gestion Des Documents D'activité Pour Une Gouvernance Documentaire Stratégique Documentaire StrategiqueCecília CabselaPas encore d'évaluation
- Chapitre 2 Le Management de La QualitéDocument10 pagesChapitre 2 Le Management de La QualitéNihed Ghaoui100% (1)
- RUP ExposéDocument23 pagesRUP ExposéAbdelkader Jaafar100% (1)
- ANA Cours - Analyser Et Décrire Un SystèmeDocument23 pagesANA Cours - Analyser Et Décrire Un SystèmeArnold KpovihouanouPas encore d'évaluation
- Chapitre IDocument17 pagesChapitre ISonia Selmani Rachdi100% (2)
- Le trading en ligne facile à apprendre: Comment devenir un trader en ligne et apprendre à investir avec succèsD'EverandLe trading en ligne facile à apprendre: Comment devenir un trader en ligne et apprendre à investir avec succèsÉvaluation : 3.5 sur 5 étoiles3.5/5 (19)
- Investir pour les débutants - Démarrer en 10 étapes facilesD'EverandInvestir pour les débutants - Démarrer en 10 étapes facilesÉvaluation : 3.5 sur 5 étoiles3.5/5 (2)
- L'analyse fondamentale facile à apprendre: Le guide d'introduction aux techniques et stratégies d'analyse fondamentale pour anticiper les événements qui font bouger les marchésD'EverandL'analyse fondamentale facile à apprendre: Le guide d'introduction aux techniques et stratégies d'analyse fondamentale pour anticiper les événements qui font bouger les marchésÉvaluation : 3.5 sur 5 étoiles3.5/5 (4)
- Le Bon Accord avec le Bon Fournisseur: Comment Mobiliser Toute la Puissance de vos Partenaires Commerciaux pour Réaliser vos ObjectifsD'EverandLe Bon Accord avec le Bon Fournisseur: Comment Mobiliser Toute la Puissance de vos Partenaires Commerciaux pour Réaliser vos ObjectifsÉvaluation : 4 sur 5 étoiles4/5 (2)
- Le Scalping est Amusant!: Partie 1: Le trading rapide avec Heikin AshiD'EverandLe Scalping est Amusant!: Partie 1: Le trading rapide avec Heikin AshiÉvaluation : 5 sur 5 étoiles5/5 (1)
- 7 Techniques Pour Augmenter Vos Revenus: Rentabilisez vos passions, Testez vos idées et Lancez votre business sans risqueD'Everand7 Techniques Pour Augmenter Vos Revenus: Rentabilisez vos passions, Testez vos idées et Lancez votre business sans risqueÉvaluation : 2.5 sur 5 étoiles2.5/5 (3)
- La comptabilité facile et ludique: Il n'a jamais été aussi simple de l'apprendreD'EverandLa comptabilité facile et ludique: Il n'a jamais été aussi simple de l'apprendreÉvaluation : 2 sur 5 étoiles2/5 (1)
- Le marketing d'affiliation en 4 étapes: Comment gagner de l'argent avec des affiliés en créant des systèmes commerciaux qui fonctionnentD'EverandLe marketing d'affiliation en 4 étapes: Comment gagner de l'argent avec des affiliés en créant des systèmes commerciaux qui fonctionnentPas encore d'évaluation
- Réussir son marketing par courriel : Communiquer - Fidéliser - MonétiserD'EverandRéussir son marketing par courriel : Communiquer - Fidéliser - MonétiserPas encore d'évaluation
- Comment Développer Votre Entreprise de Marketing de Réseau en 15 Minutes Par Jour : Rapide ! Efficace ! Fantastique !D'EverandComment Développer Votre Entreprise de Marketing de Réseau en 15 Minutes Par Jour : Rapide ! Efficace ! Fantastique !Évaluation : 4 sur 5 étoiles4/5 (8)
- Ce que vos commerciaux ne font pas et qui vous coûte des millionsD'EverandCe que vos commerciaux ne font pas et qui vous coûte des millionsÉvaluation : 4 sur 5 étoiles4/5 (2)
- Si tu n’es pas le premier, tu es le dernier: Stratégies de vente pour dominer votre marché et devancer vos concurrentsD'EverandSi tu n’es pas le premier, tu es le dernier: Stratégies de vente pour dominer votre marché et devancer vos concurrentsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Gestion de projet : outils pour la vie quotidienneD'EverandGestion de projet : outils pour la vie quotidienneÉvaluation : 5 sur 5 étoiles5/5 (2)
- Le jardin des vertueux: Riyad al-SalihinD'EverandLe jardin des vertueux: Riyad al-SalihinÉvaluation : 5 sur 5 étoiles5/5 (1)
- Les Secrets du MLM: Les Secrets des marketers de réseau compétentsD'EverandLes Secrets du MLM: Les Secrets des marketers de réseau compétentsPas encore d'évaluation
- Création d'une start-up à succès de A à Z: Réussir votre Start-up 2.0 Web et MobileD'EverandCréation d'une start-up à succès de A à Z: Réussir votre Start-up 2.0 Web et MobileÉvaluation : 3.5 sur 5 étoiles3.5/5 (4)
- Maîtriser l'Art de la Lettre de Motivation: ...et décrocher plus d'entretiens d'embaucheD'EverandMaîtriser l'Art de la Lettre de Motivation: ...et décrocher plus d'entretiens d'embaucheÉvaluation : 4.5 sur 5 étoiles4.5/5 (2)
- Agile Practice Guide (French)D'EverandAgile Practice Guide (French)Évaluation : 4 sur 5 étoiles4/5 (2)
- Le money management facile à apprendre: Comment tirer profit des techniques et stratégies de gestion de l'argent pour améliorer l'activité de trading en ligneD'EverandLe money management facile à apprendre: Comment tirer profit des techniques et stratégies de gestion de l'argent pour améliorer l'activité de trading en ligneÉvaluation : 3 sur 5 étoiles3/5 (3)
- Guide OCDE-FAO pour des filières agricoles responsablesD'EverandGuide OCDE-FAO pour des filières agricoles responsablesPas encore d'évaluation
- Direction, Alignment, Commitment: Achieving Better Results Through Leadership, First Edition (French)D'EverandDirection, Alignment, Commitment: Achieving Better Results Through Leadership, First Edition (French)Pas encore d'évaluation
- La communication professionnelle facile à apprendre: Le guide pratique de la communication professionnelle et des meilleures stratégies de communication d'entrepriseD'EverandLa communication professionnelle facile à apprendre: Le guide pratique de la communication professionnelle et des meilleures stratégies de communication d'entrepriseÉvaluation : 5 sur 5 étoiles5/5 (1)
- Le plan marketing en 4 étapes: Stratégies et étapes clés pour créer des plans de marketing qui fonctionnentD'EverandLe plan marketing en 4 étapes: Stratégies et étapes clés pour créer des plans de marketing qui fonctionnentPas encore d'évaluation