Chapitre I : Généralités sur les méthodes chromatographiques
Chapitre I :
Généralités sur les
méthodes
chromatographiques
0
� Chapitre I : Généralités sur les méthodes chromatographiques
a) 1-Définition :
b) La chromatographie est une méthode qui permet de séparer les constituants d’un
mélange en phase homogène liquide ou gazeux. C’est une technique de séparation très
puissante qui permet de travailler sur quantités allant jusqu’au µg
c) Lee terme chromatographie vient du grec «chroma =couleur» et «graphein=écrire» a été
réalisée en 1906 par le botaniste russe Mikhail Tswett.
d) Chromatographie et consistait à séparer les pigments d’une feuille d’épinard sur une
colonne en CaCO3 .
e) Tswett constatait : Tout comme les rayons lumineux d’un spectre , les différentes
composantes d’un mélange de colorants se déploient sur la colonne de carbonate de
calcium selon une loi et peuvent ȇtre analyser qualitativement et quantitativement.
f) 2-Principe des méthodes chromatographiques :
Les méthodes chromatographiques sont des méthodes de séparation mettant jeu
différents processus physicochimiques.
Historiquement,l’apparition de ces techniques remonte à 1903,date à laquelle le botaniste
russe [Link] a réalisé la séparation de pigments végétaux de la chlorophylle sur une
colonne remplie de carbonate de calcium avec de l’éther de pé[Link] séparation du mélange
en différentes bandes colorées a donné son nom à la méthode chromatographique(du grec
khroma qui signifie couleur).Alors que cette première séparation était basée sur les différeces
d’adsorption des pigments,les méthodes chromatographiques actuelles mettent à profit
différents phénomènes applicables à des substances coloeées ou [Link] évolutions ont suivi
la première expérience de [Link] parmi les plus importantes,l’apparition de :
-1938 :la chromatographie sur couches minces (Ismailov et Shraiber) ;
-1939 : la chromatographie par échanges d’ions(Samuelson) ;
-1941 : la chromatographie de chromatographiques partage liquide_liquide(Martin et
Synge) ;
-1952 : la chromatographie en phase gazeuse sur colonnes remplies(James et Martin) ;
-1959 : la chromatographie en phase gazeuse sur colonnes capillaires(Golay) ;
-1962 :la chromatographie en phase supercritique(Klesper) ;
-1968 :la chromatographie en phase liquide haute performance(Giddings et Kirkland).
1
� Chapitre I : Généralités sur les méthodes chromatographiques
L’origine de l’efficacité des techniques chromatographiques réside incontestablement dans
le fait que les constituants du mélange à résoudre entrrnt dans une suite continue d’un nombre
considérable d’équilibres avec deux phases :
_l’une stationnaire, appelée phase fixe ;
_l’autre dite mobile qui parcourt la première avec une vitesse déterminée.
Il en résulte que chaque constituant du mélange s’équilibre entre deux phases un très
grand nombre de fois et se trouve finalement entraȋné par la phase mobile à parcourir la phase
stationnaire avec une vitesse qui lui est propre.
En conséquence :
• Si le temps de migration de la phase mobile est déterminé, chaque constituant
d’un mélange parcourt au cours de l’expérience une certaine distance sur la phase
stationnaire du fait de sa vitesse [Link] l’expérience est réalisée avec une
colonne remplie de phase stationnaire,au temps t,le composé A plus rapide aura
parcouru une distance d1 supérieure à celle d2 du compsé B.
Dans ces conditions,nous venons de réaliser une chromatographie de
dé[Link] peut tout à fait imaginer qu’il est possible de récupérer les
bandes A et B en découpant la colonne(ceci a été effectivement fait dans le
passé).1
Figure 1 : Séparation de deux composés dans une colonne remplie de phase
stationnaire
• L’autre alternative consiste à fixer la distance de migration, par exemple toute la
longueur de la [Link] en résulte,alors, des temps de sortie différents pour
1
Gwenola Burgot , Jean-Louis Burgot: Méthodes instrumentales d’analyse chimique et applications
(3eéditions),Paris ,2011,p 04.
Gwenola Burgot , Jean-Louis Burgot: Méthodes instrumentales d’analyse chimique et applications
(3eéditions),Paris ,2011,p 11.
2
� Chapitre I : Généralités sur les méthodes chromatographiques
chacun des composé[Link] deuxième procéde encore appelé chromatographie
d’élution est le plus utilisé à l’heure actuelle.2
3-Pics chromatographiques :
La détection des espèces sortantes se fera sur un chromatogramme, grâce à des pics
[Link] pics ne correspondent pas à un seul point fixe mais à une
« déformation » qui tend vers un profil gaussien. Cette déformation est due aux nombreux
transferts entre la phase stationnaire3.
On définit un pic chromatographique par plusieurs éléments :
-la hauteur du pic h
-la largeur à mi-hauteur δ =2.35 σ
-la largeur à la base du pic ω=4 σ =1.7δ
Le premier pic à apparaitre sur un chromatogramme permet de déterminer le temps mot
tm ,qui est le temps que met la phase mobile pour traverser la colonne .En
chromatographie ,les différents pics correspondent , soit à des temps de rétention (temps que
mettra le composé pour traverser la colonne),soit à des volumes de rétention(volume de
phase mobile nécessaire pour éluer le composé)ou encore à des distances de rétention
(distance relative à l’apparition du pic sur le chromatogramme par rapport au début du
tracé).Il est très aisé de passer de l’un à l’[Link] toutefois dans les formules
mathématiques à ne pas mélanger temps ,distance et [Link] les formules on peut
2
Gwenola Burgot , Jean-Louis Burgot: Méthodes instrumentales d’analyse chimique et applications
(3eéditions),Paris ,2011,p 05.
3
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017, p
17.
3
� Chapitre I : Généralités sur les méthodes chromatographiques
donc extrapoler l’ensemble des formules ( qui seront ici données en temps de rétention )en
distance et en volume4.
4-Théorie des plateaux :
La colonne chromatographique est séparée en plateaux de hauteurs é[Link] le
nombre de plateaux (N) sera éleve, plus la séparation sera fine et la résolution (Rs) élevée5.
On définit ainsi plusieurs grandeurs en utilisant L la longueur de la colonne ainsi que les
paramètres des pics chromatographiques vus précédemment .
L
La hauteur équivalente à un plateau théorique HEPT=H=
N
4
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017, p
17.
5
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017, p
18.
4
� Chapitre I : Généralités sur les méthodes chromatographiques
2 t 2r 2 2
L t r tr tr
Le nombre de plateaux théoriques N= =
H σ 2
( W4 ) 2 =16 ω =5.54 δ
2 2
On peut augmenter N en réduisant le débit de la phase mobile mais l’analyse en sera plus
longue .
5-Théorie cinétique :
Ici ,on prend en compte des paramètres propres à la colonne chromatographique pour
expliquer la sé[Link] plus de la vitesse moyenne de déplacement des molécules de la
phase mobile(u),3 paramètres mathématiques entrent en jeu (A,B et C)6.
L
u= t
m
L est la longueur de la colonne.
tm est le temps mort (temps qu’un composé n’ayant aucune affinité pour la PS mettra à
traverser la colonne)
[Link] diffusion turbulente (A)
Elle dépend de la taille et de la répartition des particules de la phases [Link]
cheminement emprunté par la molécule déterminera ce facteur (en effet plusieurs chemins
étant possibles au travers de ces particules ).Plus les particules seront fines ,plus ce facteur
sera petit et l’efficacité de la colonne augmentera .
Ca facteur est nul pour une chromatographie à colonne capillaire7.
A=2.λ.d p
λ, constante de régularité du remplissage
d p ,diamètre des patticules
B
[Link] diffusion moléculaire longitudinale( U )
6
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017, p
18.
7
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017, p
18.
5
� Chapitre I : Généralités sur les méthodes chromatographiques
Elle traduit la dispersion des particules du soluté dans la colonne,celles_ci se dispersant
d’autant plus dans toutes les directions à mesure que le débit est faible.
En travaillant en hautes pressions (ex :CLHP),les débits obtenus permettent d’avoir de
bonnes séparation8.
B=2 γ .Dm
γ ,constante liée à l’espase entre les particules de remplissage
Dm ,coefficient de diffusion du soluté dans la phase mobile(en cm2.s-1)
[Link] résistance au transfert de masse(Cu)
Elle représente la résistance qui existe entre les échanges de soluté entre phase mobile et
phase [Link] le débit de la phase mobile est important ,plus C [Link]
empeche la formation de l’équilibre entre le soluté dans la PM et la PS .L’efficacite sera
donc fonction du diamètre des particules et de la diffusion du soluté.
2
d
C= p
Dm
dp ,le diamètre des particules
Dm ,coefficient de diffusion du soluté dans la phase mobile( en cm2.s-1)
[Link] de Van Deemter
8
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017, p
19.
6
� Chapitre I : Généralités sur les méthodes chromatographiques
Cette représentation mathématique permet de rassembler les termes précédemment
décrit,ceci afin d’optimiser la largeur des pics chromatgraphiques ,en modulant le débit de
la phase mobile .
La courbe de Van Deemter est la résultante des paramètres(A,B etC) qui représenteront des
constantes ,et le débit de la phase mobile (u) sera la [Link] cherchera donc à obtenir H
le plus faible possible, afin d’avoir la meilleur efficacité9.
Courbe de Van Deemter
Le débit optimal est alors obtenu au point d’inflexion de la courbe .Pour le calculer il faut
utiliser la dérivée mathématique et la résoudre .
B
H=A+ +C.u
U
H’= - B ⁄ U 2+C
B
On veut H’=0 soit - 2 +C=0
u
Après résolution on trouve uoptimal =
B
C √
[Link] de Knox (CLHP)
Le raisonnement est semblable au paragraphe précédent mais les calculs sont un peu plus la
résolution .
A B
H= 1 + +C.u
u 3 U
6. Eléments de mesures :
9
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017, p
19.
7
� Chapitre I : Généralités sur les méthodes chromatographiques
a .Rétention
i . Distribution
le soluté se répartit entre la phase stationnaire et la phase [Link] distribution se mesure
alors par le rapport des concentrations dans chacune des phases ,on obtient alors le coefficient
de distribution K10.
Cs
K=
Cm
Cs ,la concentration dans la phase stationnaire
Cm ,la concentration dans la phase mobile
[Link] et volume de rétention
Il s’agit du temps ou du volume nécessaire de phase mobile que pour qu’un composé
traverse la colonne chromatographique.
Au niveau du chromatogramme, c’est l’endroit ou’le pic chromatographique est maximal.
En connaissant le débit D de la phase mobile, on peut facilement déterminer le temps de
rétention tr grace au volume de rétention Vr.
Vr= tr.D que l’on peut extrapoler à Vm =tm.D (volume et temps mort )
On peut relier le volume de rétention aux volumes de phases mobiles et stationnaire grace au
facteur de distribution.
Vr=Vm+[Link]
Le temps (ou volume de rétention )est dépendant de plusieurs paramètres :
-La vitesse de la phase mobile
-Le coefficient de distribition du soluté
-Le rapport des volumes
On peut donc le modifier en changeant la composition de la phase mobile ou en modifiant la
température car K est dépendant de la température.
[Link] de rétention
On utilise le facteur de rétention K’ plutôt que le facteur de distribution pour s’affranchir des
paramètres géométriques de la colonne .
C s .V Vs
K’= s =K.
Cm . V m Vm
En extrapolant avec les formules précédentes :
10
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017,
p 20.
8
� Chapitre I : Généralités sur les méthodes chromatographiques
Vr
Vs= ¿
Vr Vr t r −t
K’= K . k . V = V =
¿ ¿ m
m m tm
On en déduit également
tr=tm(1+K’) et Vr=Vm(1+k’)
b.Sélectivité
le facteur de sélectivité α caractérise la distance qui sépare les sommets de deux pics11.
Kb K 'b t
α= = = ¿
K a K ' a t r −t m a
α diminue à mesure que l’on augmente la température .
Ce facteur mesure la différence de distribution thermodynamiques des deux composés.
Si ∆ ¿ )=∆ G° b-∆ G° a , on a ∆ ¿ )=-RT In α
c.Résolution
Pour avoir une bonne séparation des compsés,les pics chromatographiques doivent ȇtre les
plus distincts possibles (chevauchements minimums), ceci est mesuré grâce à la résolution
(Rs).Il a été démontré que la résolution est optimale si il y a plus de 99,8% de séparation drs
pics , ce qui correspond à une Rs ¿ 1.5.
Cela correspond à une résolution optimale ,on pourra par exemple moduler le débit (donc
le temps d’analyse) pour améliorer la résolution .Il n’est cependant pas nécessaire d’avoir une
résolution trop élevée sous peine que le temps d’analyse ne s’allonge inutilement12.
11
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017,
p 20.
12
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017,
p 20.
9
� Chapitre I : Généralités sur les méthodes chromatographiques
Mauvaise Résolution optimale ,bonne Résolution trop
séparation ,résolution trop séparation et temps élevée,temps analyse long
faible acceptable
t¿ t¿
Rs=2. =1.18.
W b +W a δ b+ δ a
Avec : - la largeur à mi-hauteur δ =2.35σ
-la largeur à la base du pic ω=4 σ =1.7 δ
On peut é galement estimer la r é solution grace à des approximations prenant en compte≤nombre de
plateaux théoriques N ,le coefficientde sélectivité α , et le facteur de rétention K’.
Approximations de purnell :Si on suppose que ωa=b
1 α
→ Rs=
4 √ Nb . 1 .
α
Si on prend en compte la moyenne du nombre de plateaux théoriques N et du facteur de
rétention K ' des composés A et B
1 α −1
→ Rs=
2 √ N.
α+ 1
.
On peut alors optimiser la séparation en :
-Augmentant N par modification de la vitesse d’élution.
-Augmentant K b par modification de la phase mobile PM.
- Diminuant α par augmentation de la température.
[Link] de charge dans une colonne
On la détermine grace à la loi de Darcy :
u
∆ P=∅ . η.L. 2
dp
∆ P,la perte de charge en Pa
∅ ,le facteur de résistance à l’écoulement
η , la viscosité en Pa.s
L , la vitesse de la phase mobile en cm.s-1
dp, le diamètre des particules en cm
[Link],porosité et débit
La colonne chromatographique est définie selon plusieurs paramètres .
10
� Chapitre I : Généralités sur les méthodes chromatographiques
Le volume de la phase mobile : Vpm=Vi(volume interstitiel)+ Vp(volume des pores)
Volume total de la colonne : VT=Vpm+Vps(volume phase stationnaire)
Volume de la colonne (si on l’apparente à un cylindre à rayon r et de longueur L ) Vc= π .
r2 .L13
V pm
La porosité de la colone : ε =
VT
Vc
Le débit de la phase mobile: D= u.ε . π . r 2 =ε .
tm
L
Avec u la vitesse de la phase mobile ( u = ) et r le rayon de colonne .
tm
7. Optimisation des performances d’une colonne :
Effet des facteurs de rétention et de sélectivité sur la résolution :
Il peut ȇtre utile de développer une relation mathématique entre la résolution d’une colonne et
les facteurs de rétention K’A et K’B de deux solutés,le facteur de sélectivité α et le nombre de
plateaux N de la [Link] ce but,on admettra que les temps de rétention des solutés A et
B sont suffisamment voisins que l’on puisse écrire14
WA=WB≈ W
L’équation 1 devient alors
Rs =¿ ¿ ¿
L’équation 2 permet d’exprimer W en termes de (tR)B et N ,qui par substitution dans
l’équation précédente,donne
13
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017,
p 21.
14
Exercices méthodologiques : Guillaume Grzych, Claire Duployez, Prépa Pharma, (2 eéditions), belgique, 2017,
p 21.
11
� Chapitre I : Généralités sur les méthodes chromatographiques
Rs=¿ ¿ ¿ ×
√N
4
La substitution dans l’équation 3 et le réarrangement des termes conduisent à une expression
de RS en fonction des fanteurs de capacité de A et de B. On écrit
K ' B− K '
×√
N
Rs = A
1+ K ' B 4
En éliminant K’A dans cette zxpression par substitution dans l’équation 4, on obtient
( α )( 1+ K ' )
√ N α −1 K 'B
Rs= (5)
4 B
Ilest suvent souhaitable de calculer le nombre plateaux théoriques requis pour obtenir la
résolution souhaitée .Une transformation directe de l’équation 5 donne
( α −1 )( )
α 1+ K ' B
N=16Rs2 2 2
(6)
K 'B
On recontre parfois des formes simplifiées des équations 5 et 6 lorsque l’on tente de séparer
très voisines rendent leur séparation [Link], lorsque KA≈ KB, l’équation 7 indique que
K’A≈ K’B=K’ et l’équation 8, que α → [Link] approximations conduisent ,par l’intermédiaire
des équations 5 et 6 ,aux formes simplifiées15
Rs¿
4 ( 1+ K ' )
√ N (α −1 ¿ K ' (9)
N=16R2s ( α −1
1
) ( 1+KK' ' )
2 2
(10)
Ou K’ est égal à la valeur moyenne de K’A et K’B .
Effet de la résolution sur le temps de rétention
Avant d’examiner en détail la signification des quatre équations qui viennent d’etre
obtenues,il est utile de développer une équation relative à une propriété connexe d’une
colonne,le temps nécessaire pour obtenir la séparation des solutés A et B , le but de la
chromatographie étant,comme on l’a mentionné précédemment ,d’obtenir la meilleure
résolution possible dans le temps le plus court .Toutefois, ces deux objectifs sont
généralement incompatibles, et un compromis entre les deux doit etre accepté16.
15
Skoog, Holler, Nieman: Principes d’analyse instrumentale (5 eéditions),Espagne ,2003,p 689.
16
Skoog, Holler, Nieman: Principes d’analyse instrumentale (5 eéditions),Espagne ,2003,p 689.
12
� Chapitre I : Généralités sur les méthodes chromatographiques
Le temps (tR)B requis pour une séparation est déterminé par la vitesse vB du soluté le plus
lent (équation 11) ; il s’éctit
v B= L
¿¿¿
En combinant cette expression avec 12 et 13,on obtient
NH ( 1+ K ' B )
(tR)B =
u
Ou’ u est la vitesse linéaire d’écoulement de la phase mobile .Quand on combine cette
équation avec l’équation 6, on obtient
3
16 R 2s H α (1+ K 'B )
( )
2
(tR)B= 2
(14)
u α −1 ( K 'B )
Paramètres qui déterminent la performance de la colonne :
Les équations 5 et 14 sont d’une utilité certaine car elles guident l’expérimentateur dans le
choix des conditions optimales pour atteindre un objectif qui se dérobe parfois : une bonne
séparation dans un temps minimum.L’examen de ces équations montre que chacue d’elles
contient trois éléments .Le premier est relié aux effets cinétiques qui conduisent à
l’élargissement des bandes, H /u ou N 1 /[Link] deuxième et le troisième dépandants.
Pour obtenir la meilleure résolution,on doit donc doubler la durée de la séparation17 .
(f)
En remplaçant H1et H2 dans l’équation 14 et en divisant le résulat d’une opération par
l’autre, on obtient
H1
¿ ¿ ¿ ¿ =¿ ¿ ¿ ¿ ×
H2
Où les indices 1 et 2 se rapportent aux hauteurs équivalentes de plateau théorique initialeset
finales .En réarrangeant les termes,on peut écrire
( 1,06 )2
−3
H2=H1¿ ¿ ¿ ¿ =8,7×10 cm 2
( 1,5 )
=4,3×10−3cm
17
Skoog, Holler, Nieman: Principes d’analyse instrumentale (5 eéditions),Espagne ,2003,p 689.
13
� Chapitre I : Généralités sur les méthodes chromatographiques
Ainsi, pour obtenir une résolution de 1,5 en 17,63 minutes sur une colonne de 30 cm,il est
nécessaire de diminuer de moitié la hauteur équivalente à un plateau théorique.
Modification de H
L’exemple 1f a montré que l’on pouvait améliorer résolution de manière significative en
diminuant la hauteur équivalente à un plateau théorique sans cependant allonger la durée de
l’expérience .Le tableau 1 indique les paramètres dont le réglage contribuera à ce
[Link] qu’une diminution des dimensions particulaires du matériau de remplissage
B
conduit à une nette amélioration de [Link] la phase mobile est un liquide, est très
u
souvent négligeable, on peut alors diminuer H en utilisant un slvant moins visqueux,ce qui
revient à augmenter le coefficient de diffusion du soluté dans la phase mobile18.
Modification du facteur de rétention :
Une séparation peut souvent etre optimisée de manière importante par une modification du
facteur de rétention K’B.L’augmentation de K’B améliore généralement la résolution, mais au
détriment de la durée d’é[Link] déterminer le domaine optimal des valeur de K’B,on
réécrit l’équation 5 sous la forme
Q K 'B
Rs=
1+ K ' B
et l’équation 15
3
( 1+ K ' B )
(tR)B=Q’ 2
( K ' B)
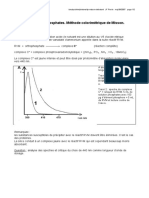
Où Q et Q’ contiennent les termes non utilisés des deux équations .La figure 2 représente un
graphique de R s /Q et ( t R ) B / Q' en fonction de K’B ,ou’ l’on a admis que Q et Q’ demeurent
pratiquement [Link] est clair qu’il faut écarer les valeurs de supéieures à10 car elles
n’entraȋnent qu’une faible augmentation de la résolution au détrment d’un allongement
considérable des durées de sé[Link] minimum de la courbe de temps d’élution
correspond à une valeur de K’B voisine de [Link] valeurs optimales de K’B sont généralement
comprises entre 1 et 5.
18
Skoog, Holler, Nieman: Principes d’analyse instrumentale (5 eéditions),Espagne ,2003,p 689-691.
14
� Chapitre I : Généralités sur les méthodes chromatographiques
Le moyen le plus fréquent d’amélioration de la résolution consiste à d’optimiser K’ .Pour
les phases mobiles gazeuses,K’ peut souvent être contrôlé par un changement de la
température de [Link] les phases mobiles liquides,des changements de composition du
solvant permettent souvent d’gir sur K’ de manièreà obtenir de meilleures sé[Link]
figure 3 montre un exemple de l’effet spectaculaire de simples changements de solvants.
Dans ce cas,de faibles variations du rapport méthannol¿ eau transforment des
chromatogrammes très peu satisfaisants (a et b) en courbes où les pics de chaque constituant
sont bien séparés(c et d).En fin de compte,le chromatogramme (c) est le meilleur puisqu’il
combine à la fois une bonne résolution et un minimum de temps19.
Modification du facteur de sélectivité :
Cependant,si α est proche de 1,l’optimisation de K’ et l’augmentation de N ne suffisent pas
à l’obtention d’une bonne séparation de deux solutés en un temps [Link] ce cas, on
doit rechercher le moyen d’accroître α tout en maintenant K’ entre 1 et [Link] options
sont possibles :par ordre de préférence décrissante, on peut (1) modifier la composition de la
phase mobile,(2) modifier la température de la colonne,(3) changer la composition de la phase
stationnaire et(4) utiliser des effets chimiques spéciaux.
Figure 2 : Effet de facteur de capaçité K’B sur la résolution Rs st sur le temps d’élution
(tR)B .On a admis que Q et Q’ restent constants lorsque K’B varie .
19
Skoog, Holler, Nieman: Principes d’analyse instrumentale (5 eéditions),Espagne ,2003,p 691.
15
� Chapitre I : Généralités sur les méthodes chromatographiques
Figure 3 : Effet de modification de solvant sur des chromatogrammes . Analytes (1)
anthraquinone-9,10 ;(2) méthyl-2-anthraquinone-9,10 ;(3) éthyl-2-anthraquinone-9,10;(4)
diméthyl-1,4-anthraquinone-9,10;(5) tbutyl-2-anthraquinone-9,10.(Document de DuPont
Biotechnology Systems,Wilmington,DE .)
La séparation de l’anisole(C6H5OCH3) et du benzène11 est un exemple de l’option [Link] une
phase mobile constituée d’un mélange de 50% d’eau et d’éthanol, K’ vaut 4,5 pour l’anisole
et 4,7 pour le benzène,tandis que α ne vaut que 1,[Link] utilisant une phase mobile aqueuse
contenant 37% de tétrahydrofuranne, les valeurs de K’ sont égales respectivement à 3,9 et 4,7
et celle de α , à 1,[Link] chevauchement des pics,importante avec le premier solvant, est
négligeable avec le second.
Dans le cas de séparations qui impliquent des acides ou des bases,une modification du pH
de la phase mobile permet souvent de faire varier α sans changement significatif de K’,ce qui
entraîne une amélioration de l’effcacité de la séparation.
Pour augmenter α tout en consevant des valeurs optimales de K’,une méthode moins
commde, mais souvent extrȇmement efficase,consiste à modifier la composistion chimique de
la phase [Link] tirer profit de cette option,la plupart des laboratoires qui effectuent
des séparations chromatographiques disposent d’un ensemble de colonnes que l’on peut
permuter facilement.
16
� Chapitre I : Généralités sur les méthodes chromatographiques
Une augmentation de la température entraȋne habituellement celle de K’,elle a cependant
peu d’effet sur les valeurs de α en chromatographie liquide-liquide et liquide-solide.L’effet de
la température peut ȇtre important, au point qu’il peut ȇtre utile de l’exploiter avant de penser
à modifier le remplissage de la colonne.
Une dernière méthode consiste à incorporer à la phase stationnaire une espèce
chimique qui complexe,ou du moins réagit avec un ou plusieurs constituants de
l’é[Link] exemple, en imprégnant l’adsorbant d’ions argent,la formation de complexes
entre les ions argent et les composés organiques insaturés améliore la séparation des
oléfines20 .
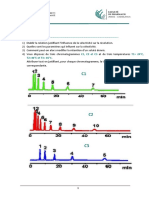
Problème général de l’élution :
La figure 4 représente les chromatogrammes hypothétiques d’un mélange de six
constituants groupés en trois paires dont les coefficients de distribution,et donc les facteurs de
capacité,sont extrȇmement différents.
Dans le chromatogramme(a),les conditions ont été ajustées pour que les facteurs de capacité
des constituants 1 et 2 (K’1 et K’2) aient des valeurs optimales comprises entre 1 et
[Link],les facteurs correspondants des autres constitants sont beacoup plus élevés que la
valeur optimale.Dès lors,les pics des constituants 5 et 6 n’apparaissent qu’après un temps
extrȇmement long ;en outre,ils sont tellement larges qu’il devient difficile de les identifier
sans ambiguїté21.
Figure 4 : Résolution de mélanges complexes par chromatographie.
20
Skoog, Holler, Nieman: Principes d’analyse instrumentale (5 eéditions),Espagne ,2003,p 691.
21
Skoog, Holler, Nieman: Principes d’analyse instrumentale (5 eéditions),Espagne ,2003,p 691-692.
17
� Chapitre I : Généralités sur les méthodes chromatographiques
Comme le montre le chromatogramme(b),on peut modifier les conditions pour optimiser
la séparation des constituants 5 et 6,mais cela entraȋne un regroupement des pics des quatre
premiers constituants à un point tel que leur résolution est insuffisante.C’est dans ce cas que
la durée totale d’élution est la plus courte .
Un troisième ensemble de conditions dans lesquelles les valeurs de K’ des composant 3 et
4 sont optimales fournit le chromatogramme(c).Une fois de plus,la séparation des deux autres
paires de composés n’est guère satisfaisante.
Le comportement illustré par la figure 4 se rencontre assez souvent lors de la résolution
de mélanges [Link] solution à ce problème consiste à faire varier les conditions qui
détermient les valeurs de K’ au cours mȇme de la séparation .Ces modifications peuvent ȇtre
effecuées de manière discontinue ou continue.
Ainsi, pour le mélange illustré par la figure 4,les conditions au début de l’élution doivent ȇtre
celles qui produisent le chromatogramme(a).Immédiatement apprès l’élution des constituants
1 et 2 ; on modifie les conditions pour se placer dans la situation optimale de séparation des
constitants 3 et 4, comme dans le chromatogramme(c).Dès la sortie des pics de ces
substances, l’étution doit se poursuivre dans les conditions utilisées pour obtenir le
chromatogramme(b).Une telle procédure permet souvent d’obtenir une bonne séparation de
tous les constituants d’un mélange en un minimum de temps.
Pour la chromatographie en phase liquide,on modifie la valeur de K’ en changeant la
composition de la phase mobile pendant l’élution(gradient de composition ou programmation
de solvant).Pour la chromatographie en phase gazeuse ,l’accroissement de la température
(programmation de température) permet d’optimiser les conditions de séparation22.
[Link]érȇt :
Les méthodes chromatographiques sont les méthodes les plus importantes de l’analyse
immédiate .Elle permettent,bien sȗr,de séparer les constituants d’un mélange plus ou moins
complexe mais là ne se borne pas leur intérȇt car elles peuvent également assurer dans
certaines conditions l’identification et la détermination quantitative des substances23.
22
Skoog, Holler, Nieman: Principes d’analyse instrumentale (5 eéditions),Espagne ,2003,p 692.
23
Skoog, Holler, Nieman: Principes d’analyse instrumentale (5 eéditions),Espagne ,2003,p 693.
18