Vous aimerez peut-être aussi
- Applications de la spectrophotomérie en phytochimie: sciencesD'EverandApplications de la spectrophotomérie en phytochimie: sciencesPas encore d'évaluation
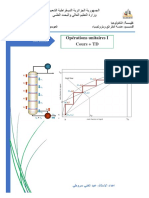
- Distillation Continue D'un Mélange BinaireDocument15 pagesDistillation Continue D'un Mélange BinaireBoudardaraPas encore d'évaluation
- La Distillation - Partie 2-: Notes de Cours: Distillation - Mme BRAKCHIDocument7 pagesLa Distillation - Partie 2-: Notes de Cours: Distillation - Mme BRAKCHIHanane KhettabiPas encore d'évaluation
- Fiche4 (Réparé)Document14 pagesFiche4 (Réparé)Belgacem ChandoulPas encore d'évaluation
- 4GPE-Diaporama Cours Réacteurs Gaz-LiquideDocument57 pages4GPE-Diaporama Cours Réacteurs Gaz-LiquideDiana Alexandra Amaya Ramirez100% (3)
- Cours de Distillation DefDocument25 pagesCours de Distillation DefFars AminePas encore d'évaluation
- Ens 2an25 Emd2 Cinetique Chimique Electrochimie16Document6 pagesEns 2an25 Emd2 Cinetique Chimique Electrochimie16okba wuni100% (1)
- 1S TP N 17 Separation D Un Melange Eau AlcoolDocument3 pages1S TP N 17 Separation D Un Melange Eau Alcoolsaber mimiPas encore d'évaluation
- Chapitre 02Document25 pagesChapitre 02Savana Bella100% (1)
- TP de Chimie Organique II - 22-23Document9 pagesTP de Chimie Organique II - 22-23Ayoub Ichfi0% (1)
- Cours DistillationDocument19 pagesCours DistillationHamzaoui AymenPas encore d'évaluation
- EXPOSé CarburantsDocument24 pagesEXPOSé CarburantsKessairi NourelhoudaPas encore d'évaluation
- Cours Kouras PDFDocument81 pagesCours Kouras PDFkarim mazizPas encore d'évaluation
- EvaporationDocument3 pagesEvaporationibtissam mikhyarPas encore d'évaluation
- Chapitre I - DTSDocument30 pagesChapitre I - DTSSalma ADAIDAPas encore d'évaluation
- Résumé Cours Chimie Verte Et Exercises-CopierDocument20 pagesRésumé Cours Chimie Verte Et Exercises-CopierSoumia Bakhta100% (1)
- Série 1Document2 pagesSérie 1slh 01Pas encore d'évaluation
- TP Chimie 2Document16 pagesTP Chimie 2khraifi RimPas encore d'évaluation
- 1636716862340 - contrel جمالDocument10 pages1636716862340 - contrel جمالGeçmiş GölgelerPas encore d'évaluation
- Mémoire Génie ChimiqueDocument104 pagesMémoire Génie ChimiqueRa NiaPas encore d'évaluation
- TP2OPUDocument16 pagesTP2OPUImene Mechkour100% (2)
- TD ElectrochimieDocument2 pagesTD ElectrochimieYahia chenna100% (1)
- Chimie de Sur2Document41 pagesChimie de Sur2Laiadhi DjemouiPas encore d'évaluation
- RH COURS 2021 BisDocument74 pagesRH COURS 2021 BisImene MechkourPas encore d'évaluation
- TP 05 Determination Du Degre Chlorometrique (Oxydo-Réduction)Document6 pagesTP 05 Determination Du Degre Chlorometrique (Oxydo-Réduction)Moume AmiraPas encore d'évaluation
- McCabe Thiele FrenchDocument21 pagesMcCabe Thiele FrenchChokri Chakiir100% (1)
- Cours Bilans MacroscopiquesDocument12 pagesCours Bilans MacroscopiquesJojo Boub100% (1)
- Cours de Rectification - Rectification ContinueDocument1 pageCours de Rectification - Rectification ContinueGiegoub EsaiePas encore d'évaluation
- Phénomènes D'adsorptionDocument2 pagesPhénomènes D'adsorptionBouchraPas encore d'évaluation
- Test 3 GP 1 PDFDocument1 pageTest 3 GP 1 PDFMourad MatmourPas encore d'évaluation
- Op Unit PDFDocument100 pagesOp Unit PDFHamstr Hamstr0% (1)
- Exo DistillationDocument1 pageExo DistillationMeng HeangPas encore d'évaluation
- 1ère SérieDocument3 pages1ère SérieAbdallah abdellaouiPas encore d'évaluation
- Réacteurs RéelsDocument71 pagesRéacteurs RéelsMohidi Mohidi100% (1)
- EMD M1 GC 2019 Kechroud Hamaodi Inal - TextMarkDocument5 pagesEMD M1 GC 2019 Kechroud Hamaodi Inal - TextMarkOmair100% (1)
- Cours SechageDocument107 pagesCours SechageDoha HaggouchiPas encore d'évaluation
- Corrigée Examen Réacteurs Polyphasiques 2022Document4 pagesCorrigée Examen Réacteurs Polyphasiques 2022ᎡᎯᏔᎯᏁᎬ ᏰᏚᎻᏞPas encore d'évaluation
- Extrait Adsorption SechageDocument44 pagesExtrait Adsorption SechageRosa100% (3)
- CGP103 Cristallisation 1 2014Document86 pagesCGP103 Cristallisation 1 2014Elie RizkPas encore d'évaluation
- TP-01-adsption-dacide-ac - Tique-Sur-Charbon-Actif - PDF Filename UTF-8''TP-01-adsption-dacide-acétique-sur-charbon-actifDocument3 pagesTP-01-adsption-dacide-ac - Tique-Sur-Charbon-Actif - PDF Filename UTF-8''TP-01-adsption-dacide-acétique-sur-charbon-actifManel 123Pas encore d'évaluation
- Chimie Controle CombustionDocument2 pagesChimie Controle Combustionimanfekryahmed100% (1)
- N I V F Fi S: Chapitre:3 Bilan de Matière Dans Les Réacteurs Idéaux GénéralitéDocument8 pagesN I V F Fi S: Chapitre:3 Bilan de Matière Dans Les Réacteurs Idéaux GénéralitéAsmaPas encore d'évaluation
- Cinétique ChimiqueDocument10 pagesCinétique Chimiquearfpower100% (2)
- TP1 Génie PétrochimiqueDocument6 pagesTP1 Génie Pétrochimiqueياسمين لقرافPas encore d'évaluation
- 1 THEORIEs DES SOLUTIONSDocument12 pages1 THEORIEs DES SOLUTIONSMira MslPas encore d'évaluation
- Cours Cinetique-Catalyse SMC5 2014Document134 pagesCours Cinetique-Catalyse SMC5 2014Yc YacinePas encore d'évaluation
- Série 2 Master PDFDocument4 pagesSérie 2 Master PDFBergoug BahaPas encore d'évaluation
- TD 1 Cryo TMPDocument1 pageTD 1 Cryo TMPMeriem Berrachedi100% (1)
- Chapitre 1 - AdsorptionDocument47 pagesChapitre 1 - AdsorptionSara Qrm100% (1)
- Fiche TD AbsorptionDocument1 pageFiche TD AbsorptionBeicha100% (1)
- TP Extraction SimpleDocument2 pagesTP Extraction SimpleOmairPas encore d'évaluation
- Phénomènes de Surface Et Catalyse HétérogèneDocument141 pagesPhénomènes de Surface Et Catalyse HétérogèneAbde TamPas encore d'évaluation
- Chap 2 Thermodynamique GAZ REELSDocument39 pagesChap 2 Thermodynamique GAZ REELSNour El Houda TebbanePas encore d'évaluation
- Distillation 07 2Document4 pagesDistillation 07 2Harifidy Niriantsoa RamanantsialoninaPas encore d'évaluation
- Transfert de Chaleur Par RayonnementDocument21 pagesTransfert de Chaleur Par Rayonnementboudad mostafaPas encore d'évaluation
- Chapitre 5 Mass TransferDocument85 pagesChapitre 5 Mass TransferJennifer Maamary100% (2)
- Calcul de Réacteurs ChimiquesDocument39 pagesCalcul de Réacteurs ChimiquesجعدبندرهمPas encore d'évaluation
- Constantes Cryoscopiques de Quelques SolvantsDocument7 pagesConstantes Cryoscopiques de Quelques SolvantsKhalil OukebdanePas encore d'évaluation
- Copie de L'Évaporation - Docx - Google DocsDocument19 pagesCopie de L'Évaporation - Docx - Google DocsMourad MatmourPas encore d'évaluation
- Thermodynamique: Les Machines Thermiques Dithermes.Document18 pagesThermodynamique: Les Machines Thermiques Dithermes.Mohamed El Hadi Redjaimia100% (5)
- Exercice Et Solution Fours ChauidierDocument46 pagesExercice Et Solution Fours ChauidierMourad MatmourPas encore d'évaluation
- Serie n1 DistillationDocument13 pagesSerie n1 DistillationMourad MatmourPas encore d'évaluation
- Cours TD AbsorptionDocument21 pagesCours TD Absorptionمحمد خشعيPas encore d'évaluation
- Introduction GeneraleDocument49 pagesIntroduction GeneraleHamdaoui douniaPas encore d'évaluation
- Opt LinDocument2 pagesOpt Linabram ibraPas encore d'évaluation
- V2 RQ X 4 QuenlennecDocument11 pagesV2 RQ X 4 QuenlennecMourad MatmourPas encore d'évaluation
- 2014-04-21 La Fiche de Lecture Et R - Sum - BoukhelifDocument8 pages2014-04-21 La Fiche de Lecture Et R - Sum - BoukhelifMourad MatmourPas encore d'évaluation
- AMDEC Moteur ThermiqueDocument46 pagesAMDEC Moteur ThermiqueMourad MatmourPas encore d'évaluation
- C55D5X DS375 CPGK RevAF1 PDFDocument3 pagesC55D5X DS375 CPGK RevAF1 PDFYacine MezianiPas encore d'évaluation
- AST09 Electromecanique Sys AutoDocument64 pagesAST09 Electromecanique Sys Autolkuima aneesPas encore d'évaluation
- 2013-11-28 Cours de Franï¿ Ais La Lettre Formelle. BoukhelifDocument13 pages2013-11-28 Cours de Franï¿ Ais La Lettre Formelle. BoukhelifMourad MatmourPas encore d'évaluation
- 2014-04-21 La Fiche de Lecture Et R - Sum - BoukhelifDocument8 pages2014-04-21 La Fiche de Lecture Et R - Sum - BoukhelifMourad MatmourPas encore d'évaluation
- Deliberation SN 2020 202master1Document21 pagesDeliberation SN 2020 202master1Mourad MatmourPas encore d'évaluation
- Chapitre2 Partie1Document8 pagesChapitre2 Partie1Mourad MatmourPas encore d'évaluation
- Cours TD AbsorptionDocument21 pagesCours TD Absorptionمحمد خشعيPas encore d'évaluation
- Ismahane Ben Ghochi: Appréciation: 14.96Document1 pageIsmahane Ben Ghochi: Appréciation: 14.96Mourad MatmourPas encore d'évaluation
- 3938 Combustion Sti2dDocument11 pages3938 Combustion Sti2dMourad MatmourPas encore d'évaluation
- Cours Thermodynamique SAKERDocument75 pagesCours Thermodynamique SAKERfary047Pas encore d'évaluation
- Devoir SDocument3 pagesDevoir SMourad MatmourPas encore d'évaluation
- bioile ملف مهمجدا كثياDocument249 pagesbioile ملف مهمجدا كثياMourad MatmourPas encore d'évaluation
- Série de TD 3 Et 4Document4 pagesSérie de TD 3 Et 4Mourad MatmourPas encore d'évaluation
- DevoirsDocument3 pagesDevoirsMourad MatmourPas encore d'évaluation
- Série de TD 3 Et 4Document4 pagesSérie de TD 3 Et 4Mourad MatmourPas encore d'évaluation
- Chapitre IVDocument18 pagesChapitre IVMourad MatmourPas encore d'évaluation
- Chapitre IVDocument18 pagesChapitre IVMourad MatmourPas encore d'évaluation
- Mémoire PDFDocument68 pagesMémoire PDFMourad MatmourPas encore d'évaluation
- Développement de Méthodes Analytiques Par LC-MS/MS Pour La Caractérisation de L'activité Et de L'expression Des CYP450s Chez L'humainDocument141 pagesDéveloppement de Méthodes Analytiques Par LC-MS/MS Pour La Caractérisation de L'activité Et de L'expression Des CYP450s Chez L'humainMourad MatmourPas encore d'évaluation
- Exercice - Hint Software - 2Document4 pagesExercice - Hint Software - 2Mohamed El AssaliPas encore d'évaluation
- TP RedresssementDocument9 pagesTP RedresssementKereme JulienPas encore d'évaluation
- PSI PHYSIQUE E3A 1 2010.extrait PDFDocument4 pagesPSI PHYSIQUE E3A 1 2010.extrait PDFLOUkmen BelPas encore d'évaluation
- Résumé Systemes Monophases Et Triphases-WatermarkDocument6 pagesRésumé Systemes Monophases Et Triphases-WatermarkChristian Bernard LéonPas encore d'évaluation
- Vishay Selection Guide PDFDocument105 pagesVishay Selection Guide PDFRoberto RedinPas encore d'évaluation
- TD Equations DifferentiellesDocument27 pagesTD Equations DifferentiellesIbrahim HARBPas encore d'évaluation
- Convertisseur 12v CC Vers 220VAC 160 WDocument11 pagesConvertisseur 12v CC Vers 220VAC 160 WguiguessPas encore d'évaluation
- Cours DiagraphiesDocument36 pagesCours DiagraphiesAkrem Hkimi100% (5)
- Exercices en ElectrotechniqueDocument5 pagesExercices en ElectrotechniqueNaruto100% (1)
- Machines Fluidiques Partie 2Document36 pagesMachines Fluidiques Partie 2Ayoub OukhalekPas encore d'évaluation
- Athénée Royal Du Condroz Jules Delot: CineyDocument23 pagesAthénée Royal Du Condroz Jules Delot: CineytchokoPas encore d'évaluation
- CC Energie Solaire PV - Master 2 IEA - 110521Document1 pageCC Energie Solaire PV - Master 2 IEA - 110521Aymard MougouPas encore d'évaluation
- Chapitre 02Document21 pagesChapitre 02Moustapha NdourPas encore d'évaluation
- Exercices Energie OndeDocument5 pagesExercices Energie OndeHaroun Amir100% (1)
- Psi Physique CCP 2 2012.enonceDocument14 pagesPsi Physique CCP 2 2012.enoncePenda NiassPas encore d'évaluation
- Exo2 Sujet CombustionSanteDocument4 pagesExo2 Sujet CombustionSanteAnge Vianney KonePas encore d'évaluation
- THermique 21 30Document10 pagesTHermique 21 30ndn NgondzaPas encore d'évaluation
- CR TP 3 EnergieDocument7 pagesCR TP 3 EnergieAbdel LatifPas encore d'évaluation
- ArduinoDocument16 pagesArduinokoumaPas encore d'évaluation
- Diodes À Jonction PNDocument15 pagesDiodes À Jonction PNanes bendjemaiPas encore d'évaluation
- Chapitre 5Document16 pagesChapitre 5John-sun LalyPas encore d'évaluation
- Appareil de CommandeDocument24 pagesAppareil de CommandeLebel NkogoPas encore d'évaluation
- CoursSolEPF2010 P4Document36 pagesCoursSolEPF2010 P4Amal LouanatePas encore d'évaluation
- Fax Phy TCD PDFDocument40 pagesFax Phy TCD PDFDeutou0% (1)
- Cours Pompes Et Stations de PompageDocument52 pagesCours Pompes Et Stations de Pompagepapa abdoul sowPas encore d'évaluation
- Tarik AyoubDocument112 pagesTarik AyoubAnonymous A13Lxz2hTPas encore d'évaluation
- Polycopie ElectroniquedepuissanceDocument69 pagesPolycopie ElectroniquedepuissanceSalim BouaziziPas encore d'évaluation
- Travaux Diriges N°3 EnergieDocument4 pagesTravaux Diriges N°3 EnergieLuse AngePas encore d'évaluation
- Chapitre2 Le Potentiel ElectriqueDocument14 pagesChapitre2 Le Potentiel ElectriqueHamza BoutlihPas encore d'évaluation
- Chap3-Caractéristiques GéométriquesDocument18 pagesChap3-Caractéristiques GéométriquesMiSs MåissaPas encore d'évaluation