Académique Documents
Professionnel Documents
Culture Documents
Chromato Liquide 2007
Chromato Liquide 2007
Transféré par
Med KassiouiTitre original
Copyright
Formats disponibles
Partager ce document
Partager ou intégrer le document
Avez-vous trouvé ce document utile ?
Ce contenu est-il inapproprié ?
Signaler ce documentDroits d'auteur :
Formats disponibles
Chromato Liquide 2007
Chromato Liquide 2007
Transféré par
Med KassiouiDroits d'auteur :
Formats disponibles
r Jean-Louis CUQ -
page 0
CHROMATOGRAPHIE LIQUIDE
Professeur Jean-Louis CUQ
r Jean-Louis CUQ -
page 1
LA CHROMATOGRAPHIE LIQUIDE
TABLE DES MATIERES
I. HISTORIQUE II. PRINCIPE DE LA CHROMATOGRAPHIE III. CLASSIFICATION DES METHODES CHROMATOGRAPHIQUES 1. Selon la nature des phases 2. Selon la nature des phnomnes mis en jeu 3. Selon la technique mise en jeu IV. THEORIE DE BASE - GRANDEURS FONDAMENTALES Coefficient de distribution 1. Grandeurs de rtention tR, VR, k' 2. Slectivit d'une colonne 3. Efficacit d'une colonne N, HEPT, Equations de Van Deempter , Knox, Giddings, Nef 4. Rsolution Rs 5. Temps d'analyse Nef / tR 6. Elution linaire 7. Perte de charge P V. OPTIMISATION DES CONDITIONS D'UNE ANALYSE 1. Optimisation de la rsolution 1) Action sur 2) Action sur k' 3) Action sur l'efficacit 1. Influence de la vitesse de la phase mobile 2. Influence du diamtre des particules 3. Influence de la colonne (longueur et diamtre) 4. Influence de l'injection (position, volume, injecteur) 2. Optimisation du temps d'analyse 1) Optimisation de NeF/tR 2) Pression et temps d'analyse 3. Optimisation de la perte de charge 4. Conclusion 5. Dtermination graphique VI. DIFFERENTS TYPES DE CHROMATOGRAPHIE 1. La chromatographie d'adsorption 1) Thermodynamique de l'adsorption 2) Adsorbants utiliss en CLS 6 7 9 10 12 12 12 13 14 14 3 4 5
20 22 23 24 25 26 26 28
r Jean-Louis CUQ -
page 2
1. Adsorbants de type I - silice ou alumine 2. Adsorbants de type III - Phases greffes polaires 3. Adsorbants de type IV - Phases greffes apolaires a) mcanisme de rtention b) influence de la phase mobile - lution en mode isocratique - lution en mode gradient c) augmentation de k' (fixation covalente, appariement d'ions) 2. La chromatographie de partage 1) Phases stationnaires 2) Phase mobile 3. La chromatographie d'change d'ions 1) Principaux types de groupements fonctionnels 2) Principaux types de matrice 3) Influence du pH de la phase mobile 4) Influence du type et de la concentration en contre ion 5) Effet de la temprature et de solvants organiques 6) Analyse avec suppresseur de phase 4. La chromatographie d'exclusion 5. La chromatographie chirale 6. La chromatographie d'affinit VII. EQUIPEMENTS UTILISES EN CHROMATOGRAPHIE 1. Dispositifs dobtention de gradients 2. Les pompes 3. Les injecteurs 4. La colonne 5. Les particules support de phase stationnaire 6. La nature des phases stationnaires 7. Les connexions 8. Les dtecteurs VIII. QUANTIFICATION 1. Mesure de laire par triangulation ou au moyen dintgrateurs 2. Mesure des coefficients de rponse 3. Influence des conditions opratoires sur lanalyse quantitative IX. TRANSPOSITION COLONNE - COUCHE MINCE X. COMPARAISON CHROMATOGRAPHIE LIQUIDE CHROMATOGRAPHIE GAZ XI. EXERCICES ET CORRIGES
28 31 32 33 35 39 43 46 48 48 50 51 51 52 52 58 60 62 63 65 66 67 67 69 69 70 76 76 77 78 78 79 80
r Jean-Louis CUQ -
page 3
I. HISTORIQUE La chromatographie est une science trs ancienne. Ainsi on trouve la premire rfrence d'un processus chromatographique dans l'Ancien Testament qui fait mention de proprits adsorptives de certaine varit de bois pour adoucir de l'eau amre. Ce sont les tanins hydrophobes qui constituaient une phase stationnaire apolaire (chromatographie dadsorption) Aristote dcrit les proprits que possdent certaines terres pour purifier l'eau de mer. Les phnomnes mis en jeu relvent ici de lchange dions. Au XVIme sicle, le strasbourgeois BRUNSWIG purifiait de l'thanol en faisant passer la vapeur travers une ponge imprgne d'huile d'olive et ralisait une exprience de chromatographie gaz-liquide 400 ans avant la "dcouverte" de cette technique. Toutefois, ce n'est qu'au dbut du XXme sicle, plus prcisment le 21 mars 1903, que naquit la chromatographie. Ce jour-l TSWETT, botaniste d'origine russe, prsenta Varsovie son premier article sur une nouvelle sorte de phnomne d'adsorption et son application l'analyse biochimique. Il y dcrivait la formation de zone colores lors de l'lution par l'ther de ptrole de pigments vgtaux dans une colonne remplie de carbonate de calcium. L'origine du mot chromatographie vient peut-tre de cette sparation de composs colors puisque CHROMA () en grec, signifie couleur. Puis les travaux de TSWETT tombrent dans l'oubli pendant une vingtaine d'annes et il faudra attendre 1931 la publication de KUHN et LEDERER sur la sparation des isomres du carotne et de la xanthophylle pour assister au dveloppement de la chromatographie en tant qu'outil analytique. A partir de cette date, la chromatographie prit son vritable essor - 1934, premier livre de ZECHMEISTER et CHOLNOKY, qui par son succs, contribua vulgariser la chromatographie. - 1938, KUHN reut le Prix Nobel. Livre La chromatographie en phase mince par IZMAILOV et SCHRAIBER Chromatographie d'change d'ions sur zolithes. - 1941, La chromatographie de partage, MARTIN-SYNGE - 1944, La chromatographie sur papier, CONSDEN-GORDON, MARTIN - 1948, Prix Nobel TISELIUS (premier dtecteur optique par mesure du changement d'indice de rfraction, premier systme de gradient d'lution) - 1952, Prix Nobel MARTIN et SYNGE MOORE et STEIN Chromatographie d'change ionique (Prix Nobel) - 1959, Chromatographie sur gel (PORATH, FLODIN) - 1969, Avnement de la chromatographie liquide moderne (HPLC, FPLC)
r Jean-Louis CUQ -
page 4
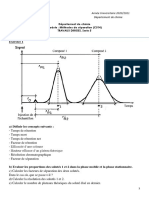
II. PRINCIPE DE LA CHROMATOGRAPHIE Il s'agit de la ralisation d'un tri entre les diffrentes espces molculaires d'un mlange. On va ainsi forcer toutes les molcules effectuer un parcours commun parsem d'obstacles : certaines espces le franchiront aisment d'autres auront plus de difficults. A l'arrive, il y aura chelonnement. Pour entraner les molcules il faut les vhiculer dans un fluide : la phase mobile qui peut tre soit un liquide soit un gaz. L'obstacle franchir, qui ne doit pas tre entran par la phase mobile, doit tre fixe et produire des effets reproductibles : il constitue la phase stationnaire. Cette phase stationnaire, le plus souvent emprisonne dans une colonne, peut tre un solide ou un liquide immobilis sur un solide. La sparation idale est schmatise sur la Figure 1.
application dveloppement rponse
temps
Figure 1. Sparation chromatographique idale
Les facteurs qui contrlent la sparation sont surtout d'ordre thermodynamique: la rtention de l'chantillon sur la colonne est donc contrle thermodynamiquement. Il est ncessaire quil y ait interaction entre lanalyte et la phase stationnaire. En pratique la sparation idale n'est pas ralisable. Ainsi il se produit un largissement des bandes en raison surtout des phnomnes de diffusion. Les facteurs qui contrlent la diffusion sont surtout de nature cintique et peuvent tre considrs indpendamment des facteurs thermodynamiques qui influencent la rtention (figure 2).
application dveloppement rponse
temps
Figure 2. Sparation chromatographique relle (non idale)
On peut classer les mthodes chromatographiques de trois manires: - selon la nature des phases - selon la nature des phnomnes mis en jeu dans la sparation - selon la technologie mise en oeuvre.
r Jean-Louis CUQ -
page 5
III. CLASSIFICATION DES METHODES CHROMATOGRAPHIQUES III.1. Classification selon la nature des phases Selon la nature de la phase mobile on distingue: - la chromatographie en phase liquide - la chromatographie en phase gazeuse - la chromatographie en phase supercritique Selon la nature de la phase stationnaire on distingue: - la chromatographie gaz / solide - la chromatographie gaz / liquide - la chromatographie liquide / solide - la chromatographie liquide / liquide CPL CPG CPS CGS CGL CLS CLL
La chromatographie en phase supercritique est une chromatographie dans laquelle la phase mobile est un gaz comprim ( sa temprature et sa pression critique) ou une phase liquide / vapeur de mme densit (au-del de la temprature critique, le gaz ne peut tre liqufi quelle que soit sa pression). Avec cette phase mobile, le plus souvent lanhydride carbonique, la dure de lanalyse est de 5 10 fois plus courte quavec les phases mobiles liquides. De viscosit trs faible, cette phase mobile permet un couplage facile avec des dtecteurs comme le spectromtre de masse ou le dtecteur par mesure de la diffusion de la lumire. III.2. Classification selon la nature des phnomnes mis en jeu Cette classification repose sur la nature de la phase stationnaire et son interaction avec les molcules sparer. On distingue ainsi: III.2.1. La chromatographie d'adsorption C'est la mthode la plus ancienne et "encore" la mieux connue. La sparation entre les molcules est fonde sur le processus rpt d'adsorption et dsorption par la phase stationnaire. Dans les premires tudes les phases stationnaires modles dtudes taient la silice et la cellulose. En consquence, les phnomnes tudis correspondaient essentiellement des interactions de type liaison hydrogne. Dautres phases ont t utilises depuis avec des mcanismes dchange impliquant des liaisons Van der Waals, hydrogne et des interactions hydrophobes (cf chapitre VI). Il s'agit de chromatographie liquide-solide (CLS) III.2.1. La chromatographie de partage La sparation est fonde sur les diffrences de solubilit des molcules sparer entre phase mobile et phase stationnaire liquide (phase qui imprgne ou qui est greffe sur un solide). Il s'agit alors de chromatographie liquide-liquide (CLL). On multiplie ici les partages entre phases de la mme faon que si l'on disposait de plusieurs milliers d'ampoules dcanter contenant deux solvants non miscibles. III.2.3. La chromatographie d'change d'ions La phase stationnaire est un solide la surface duquel se trouvent des groupements ioniss : changeur d'ions. Un solut ionis de charge oppose se trouvera d'autant plus retenu que sa charge (oppose celle
r Jean-Louis CUQ -
page 6
de la phase stationnaire) sera plus forte. Cest la loi de COULOMB qui rgit ces changes de forte nergie (40 80 kJ.mole-1) : q . q' 1 F= . 2 . 0 d La phase stationnaire solide est gnralement poreuse et renferme dans ses pores une phase liquide qui peut jouer un rle trs important dans les sparations. On peut ainsi parler pour ce type de chromatographie de CLS mais aussi de CLL. III.2.4. La chromatographie d'exclusion encore qualifie de chromatographie de permation ou de filtration sur gel La phase stationnaire est un solide poreux dont la dimension des pores est voisine de celle des molcules sparer : celles qui sont trop volumineuses pour pntrer dans les pores sont exclues de la phase stationnaire et sont lues les premires. Ici encore on peut parler indiffremment de CLS ou de CLL. III.2.5. La chromatographie d'affinit Trs utilise par les biochimistes, elle consiste fixer par exemple une enzyme sur la phase stationnaire de faon complexer slectivement les substrats correspondants (ou vice versa). Il sagit l dune association entre une molcule polyfonctionnelle et une phase stationnaire comportant des sites striquement dfinis et de capacit dchange multiple. Il sagit souvent dun systme coopratif dinteractions diffrentes (ioniques, hydrophobe, Van der Waals, hydrogne), liaisons parfaitement positionnes dans lespace. Il est bon de rappeler quil ne peut pas exister de phase stationnaire inerte . Quelle que soit sa structure elle sera toujours capable de donner des interactions dun type donn. Dans la mesure o le systme requiert un niveau dinteraction le plus faible possible, cest par les choix judicieux des phases mobiles et stationnaires quil sera possible dy parvenir. III.2. Classification selon la technique mise en jeu Selon la technologie mise en jeu on distingue: - la chromatographie sur colonne - la chromatographie de surface (chromatographie sur papier ou chromatographie sur couche mince). IV. THEORIE DE BASE - GRANDEURS FONDAMENTALES En chromatographie en phase liquide, les sparations sont bases sur la diffrence de distribution des espces entre deux phases non miscibles l'une stationnaire (particules solides imprgnes ou non d'un liquide), l'autre mobile (liquide). Pour un systme chromatographique donn, le coefficient de distribution K (ou coefficient de partage) est dfini par:
K =
Cs Cm
Cs : concentration du solut dans la phase stationnaire Cm : concentration du solut dans la phase mobile.
r Jean-Louis CUQ -
page 7
Si les molcules d'un mlange ont des affinits diffrentes pour chacune des deux phases, il apparat des diffrences entre deux vitesses de migration d'o possibilit de sparation. La migration sera d'autant plus lente que l'affinit de la molcule pour la phase stationnaire sera grande. Rappelons que la solubilit dun analyte dans une phase mobile donne passe dabord par la possibilit dtablissement dchanges (liaisons). Sans change il ny a pas de solubilit possible et deux phases apparaissent. Une bonne sparation en chromatographie liquide implique : 1. que les divers constituants du mlange soient retenus sur la colonne, donc prsentent une affinit pour la phase stationnaire suffisante pour qu'ils apparaissent dans l'effluent aprs un volume suprieur au volume interstitiel de la colonne 2. que les diffrents pics soient bien spars, ce qui pour deux pics successifs dpend de la distance sparant les sommets et leur largeur. 3. que l'analyse soit aussi rapide que possible. IV.1. GRANDEURS DE RETENTION Si la quantit inject est petite on obtient pour chaque compos lu un pic symtrique et gaussien (figure 3). On dfinit ainsi les paramtres suivants : IV.1.1. Le temps de rtention (tR) C'est le temps d'lution au maximum du pic mesur partir de l'injection. Un chromatogramme type est schmatis, avec les paramtres principaux dvaluation, sur la figure 3.
O,607 0,5 injection point mort
O,607 h 4
0,5 h
0 V M volume mort
largeur du pic la base
volume de rtention ou temps de rtention temps de rtention rduit ou temps de rtention rduit
figure 3. Principaux paramtres dun chromatogramme IV.1.2. Le volume de rtention (VR) Connaissant le dbit D de la phase mobile, suppos maintenu constant, on dfinit le volume de rtention VR = tR . D = tR . v. s v : vitesse linaire de la phase mobile s = s'. avec s' : section de la colonne VM = to . D s : section rduite de la colonne et = VM / VT (porosit)
r Jean-Louis CUQ -
page 8
VR reprsente, l'talement prs, le volume de phase mobile ncessaire pour luer chaque compos. VM est le volume de phase mobile dans la colonne. tR et VR sont des grandeurs caractristiques d'un compos (pour une colonne donne, un luant donn et des conditions exprimentales fixes) qui servent l'analyse qualitative d'un mlange. Les espces non retenues par la phase stationnaire apparaissent dans l'effluent aprs le temps to correspondant l'coulement du volume interstitiel de la colonne ou volume de phase mobile VM contenu dans la colonne. Le volume de rtention VR est reli directement au coefficient de distribution K par la relation: VR = VM + K VS VS : volume de la phase stationnaire (ou masse ou surface spcifique selon les units de K) Cette relation ne s'applique que dans le cas d'lution linaire c'est--dire quand K varie linairement avec la concentration du compos dans chaque phase. IV.1.3. Le facteur de capacit k' (ou facteur de rtention) Pour s'affranchir des paramtres gomtriques de la colonne, on utilise, pour caractriser la rtention d'un compos le facteur de capacit. En effet si K varie non linairement avec la concentration en compos dans chaque phase, un pic asymtrique se produit et VR varie avec la concentration du compos. Si on considre une petite section de bandes de longueur dx le rapport des quantits du compos dans les deux phases de cette section est le facteur de capacit de la colonne k'.
dx concentration phase mobile phase stationnaire support de phase distance x
q k' = q s = Cs .As .dx = Cs .Vs = K Vs CM .AM .dx CM .VM M VM AS et AM surface des sections de la phase stationnaire et de la phase mobile. k' est le paramtre le plus important en chromatographie liquide. De l'quation VR = VM + K VS on tire
Vs = VR - VM K d'o :
r Jean-Louis CUQ -
page 9
k' = K .
VR - VM VR - VM tR - t0 = = K . VM VM t0
Le temps et le volume de rtention sont lis au facteur de capacit par les relations :
tR = to (1 + k')
IV.2. SELECTIVITE DUNE COLONNE
VR = VM (1 + k')
Pour caractriser la distance sparant les sommets de deux pics on utilise le facteur de slectivit : = tR2 - t0 tR1 - t0
Il sagit du rapport des temps de rtention rduits
k' =
tR - t0 V soit tR - t0 = k'. t0 et k' = K . s t0 VM = k' 2 K = 2 k' 1 K1
to et VM ne dpendent que de la colonne Le facteur de slectivit mesure la diffrence de distribution thermodynamique des deux composs. On dmontre que si (Go) = G2 - G1 (diffrence des nergies libres de distribution des deux composs) on a : (G) = - RT ln IV.3. EFFICACITE DUNE COLONNE. Nombre de plateaux thoriques et de plateaux efficaces L'efficacit d'une colonne chromatographique, dont dpend l'talement des pics, est mesure, pour chaque compos, par le nombre de plateaux thoriques N de la colonne. Cette thorie est ne de la recherche d'un modle statique permettant de dcrire le fonctionnement d'une colonne chromatographique comme celui d'une colonne distiller. Au lieu de considrer le dplacement rel, continu de la phase mobile, on admet que celle-ci progresse par sauts successifs et se met en quilibre avec la phase stationnaire entre deux transferts, ce qui permet de dcouper fictivement la colonne en un certain nombre de zones dans lesquelles les quilibres sont raliss et que l'on appelle plateaux thoriques. On peut ainsi calculer le profil de rpartition des espces, de proche en proche, aprs chaque transfert et dans chaque plateau. Une colonne est alors dite comporter N plateaux thoriques si, dans des conditions donnes, elle a la mme efficacit qu'une colonne fictive de N plateaux thoriques. La thorie des plateaux tablit qu'aprs un certain parcours dans la colonne, les pics d'lution peuvent tre assimils des courbes de Gauss dont l'cart type (exprim en unit de temps) est li au nombre de plateaux thoriques parcourus par la relation : N = tR
2
Les caractristiques gomtriques de la courbe de Gauss (figure 3) permettent de calculer, pour un solut donn, N partir du chromatogramme.
r Jean-Louis CUQ -
page 10
N = 16. tR
largeur du pic la base: distance entre les points d'intersection des tangentes au point d'inflexion avec la ligne de base et largeur du pic mi-hauteur. Pour comparer entre elles des colonnes de diffrentes longueurs on dfinit la hauteur quivalente un plateau thorique. HEPT = L N
= 5,54. tR
L longueur de la colonne.
En HPLC les HEPT sont comprises entre 0,001 et 1 mm. Dans la thorie des plateaux, la HEPT (= L . 2 / t2R) dduite de la variance n'apparat que comme une mesure globale de l'influence de tous les paramtres exprimentaux. Divers modles ont t labors pour prciser les divers paramtres intervenant dans l'largissement des pics. Ainsi l'talement d'une bande de solut a trois origines : - la dispersion par diffusion axiale - l'existence de chemins multiples dus au remplissage (htrognit d'coulement) - la rsistance au transfert de masse dans chacune des 2 phases.
largeur initiale de bande diffusion axiale talement
Htrognit d'coulement
rsistance au transfert de masse dans la phase mobile talement talement dans la phase stationnaire
Principales origines de ltalement dun pic danalyte en CSL Ainsi la HEPT totale est la somme de termes dans lesquels les variances seront gales la variance totale.
HEP T = HEP T i
i
Ainsi la thorie cintique conduit l'quation de VAN DEEMTER
HEP T = X + Y + Z. v v
v vitesse rduite = vitesse linaire de la phase mobile.
talement
r Jean-Louis CUQ -
page 11
X est l'influence de la diffusion turbulente due aux htrognits dans l'coulement. Il existe plusieurs chemins possibles pour la phase mobile. Cet effet sera d'autant moins prononc que les particules seront de mme forme (sphrique) et de mme dimension (do la ncessit dutiliser de telles phases).
Y / v est l'influence de la dispersion des molcules par diffusion longitudinale; Y / v est proportionnel au coefficient de diffusion du solut dans la phase mobile DM. facteur de tortuosit (> 1) Y = 2 DM Y / v diminue quand v augmente. Comme DM est faible en milieu liquide, Y / v est gnralement ngligeable.
Zv est l'influence de la rsistance au transfert de masse. Z peut tre diminu en rduisant les distances parcourir par le solut dans chaque phase (diminution du diamtre des particules). Cette approche est surtout utilise en chromatographie phase vapeur. En chromatographie liquide on utilise souvent l'quation de KNOX
HEP T = X. v
1 3
Y v'
+ Z. v '
v' vitesse rduite
v'= v . dp DM
DM dp
coefficient de diffusion du solut dans la phase mobile diamtre des particules HEPT' = H
dp
L'quation de GIDDINGS, base sur les thories du cheminement alatoire conduit une expression analogue pour la HEPT.
HEPT = X + Y + Z.v + v
1 1 X
+1Z .v M
2 2
Au concept du nombre de plateaux thoriques, on peut associer la notion de plateaux efficaces :
N ef = k' 1 + k'
2
.N
N ef =
16. t R- t 0
2
N ef =
5,54 t R - t 0
2
soit
soit encore
Ce nombre de plateaux efficaces permet d'apprcier de manire plus concrte la vritable efficacit de sparation de la colonne. En effet, si k' est petit, la sparation peut tre impossible mme avec N grand. La capacit de pics dune colonne est dfinie par : N . ln t 16 t
n=1+
N : nombre de plateaux thoriques
r Jean-Louis CUQ -
page 12
t et t temps de rtention du premier et du dernier pic du chromatogramme. Il est ncessaire que n soit suprieur au nombre de coposs pour que la rsolution soit correcte. HERMAN (1985) a montr quavec n = 60 on a 90 % de probabilit de sparer 9 composs.
IV. 4. RESOLUTION
La rsolution Rs entre deux pics est dfinie par la relation :
Rs = 2 . t R1 - t R2
1 + 2
t R2- t
2
R1
Plus Rs est grand, meilleure est la sparation. La sparation est complte quand Rs = 1 (2 % de recouvrement des 2 pics) En supposant 2 = 1, on a :
Rs =
k' 2 - 1 1 . . . 4 1 + k' 2
N2
IV.5. TEMPS D'ANALYSE. (Nombre de plateaux efficaces par unit de temps)
t R = ( 1 + k' ) . L v
v vitesse linaire de la phase mobile. Comme HEPT = L / N
t R = N .( 1 + k' ) .
2
et
to = L / v
HEPT v
N ef =
k' .N 1 + k'
do ( division )
N ef tR = v . HEPT k'
2 3
1 + k'
IV.6. ELUTION LINEAIRE
Dans le cas d'une lution linaire, le coefficient de distribution varie linairement avec la quantit injecte. A une temprature donne, la courbe CS en fonction de CM est l'isotherme d'adsorption. Cet isotherme peut tre linaire, concave ou convexe (figure 4). En gnral en chromatographie liquide la sensibilit des dtecteurs est suffisante pour permettre l'injection de faibles quantits et l'on se trouve dans la partie linaire de l'isotherme. La pente est gale CS = K . CM
r Jean-Louis CUQ -
page 13
au coefficient de distribution K.
isotherme
isotherme
isotherme
C
rponse du dtecteur
rponse du dtecteur
CM
rponse du dtecteur
CM
t t
R
t t
R
t t
R
quantit injecte
quantit injecte
quantit injecte
Figure 4. Influence de la forme de l'isotherme d'adsorption sur la gomtrie du pic et sur le temps de rtention.
IV.7. PERTE DE CHARGE DANS UNE COLONNE
(Loi de DARCY)
P =
dp K = . 180
o 2
Lv Ko
avec
1 -
3 2
P L v K dp
perte de charge (en barye = 10-6 bars) viscosit (poise) longueur de la colonne (cm) vitesse de la phase mobile (cm.s-1) constante de permabilit (cm2) diamtre des particules (cm) porosit interstitielle (volume interstitiel de la colonne/volume total).
VERILLON et al., International Laboratory, July 1992, 29-35 proposent la loi suivante :
r Jean-Louis CUQ -
page 14
P = 400 .
F.L . d 2 . d2 p c
avec P en MPa F dbit en ml.min-1 L longueur de la colonne en cm viscosit en cP dp diamtre des particules en m et
dc diamtre de la colonne en mm
V. OPTIMISATION DES CONDITIONS D'UNE ANALYSE L'idal est d'obtenir une rsolution leve en un temps trs court. On peut envisager d'augmenter la longueur de la colonne (la vitesse d'analyse va augmenter ainsi que la perte de charge). On peut diminuer le diamtre des particules (HEPT diminue, mais la perte de charge augmente). On peut augmenter la vitesse de la phase mobile (HEPT augmente, la pression aussi). V.1. OPTIMISATION DE LA RESOLUTION Il faut augmenter Rs Rs =
k' 2 - 1 1 . . . 4 1 + k' 2
N2
ou
Rs =
1 - 1 1 . Nef 2 4
Rs doit tre > 1 V.1.1. Action sur la slectivit = tR2 - t0 tR1 - t0
La rsolution augmente quand augmente. La sparation n'est possible que si 1. Le nombre de plateaux efficace (Nef) ncessaires pour obtenir une rsolution donne en fonction de diminue trs vite quand > 1 (tableau 1). Cette forte influence de justifie l'utilisation de phases stationnaires ayant des groupements fonctionnels adquats. La slectivit peut tre modifie : - en changeant la nature et la composition de la phase mobile ce qui induit des effets secondaires (pH ionisation etc...) - en changeant la nature de la phase stationnaire - en modifiant la temprature.
r Jean-Louis CUQ -
page 15
Tableau 1. Variation du nombre de plateaux efficaces en fonction de pour obtenir Rs = 1 ou Rs = 1,5 RS = 1 1,01 1,05 1,10 1,15 1,20 1,25 160 000 6 800 1940 940 575 400 Nef RS = 1,5 360 000 15 700 4 360 2 110 1 300 900
V.1.2. Action sur le facteur de capacit k' (nature de phase stationnaire surtout) Si k' = 0 les deux soluts sont lus simultanment et aprs coulement du volume interstitiel. La rsolution augmente quand k' augmente , mais de moins en moins vite car k' tend rapidement vers 1. k'+1 Le temps d'analyse augmente beaucoup plus rapidement que k' car on a :
t R2 = 1 + k' 2 .
L longueur de la colonne et v vitesse linaire rduite. (si k' augmente de 5 10, Rs augmente de 9 %, le temps d'analyse de 80 %). V.1.3. Action sur l'efficacit Doubler N multiplie Rs par 1,41. Il n'y a pas de limite l'accroissement de Rs avec N. Si on augmente N en doublant la longueur de la colonne, on double tR car tR = L / v .(1+k'). Il faut diminuer HEPT. V.1.3.1. Influence de la vitesse de la phase mobile sur HEPT L'quation de VAN DEEMTER (ou de KNOX) permet de suivre les variations de HEPT en fonction de la vitesse linaire de la phase mobile.
HEPT CPG
L v
CPL
v
Cependant, dans un certain domaine de vitesse de la phase mobile, la HEPT dtermine exprimentalement vrifie la relation de SNYDER.
HEPT = Avn
r Jean-Louis CUQ -
page 16
n gnralement compris entre 0,2 et 0,7. A constante pour une colonne donne; v : vitesse linaire rduite de la phase mobile; n : coefficient fonction de la sparation tudie.
140
dp = 12 m dp = 5 m
120
100
80
60
40
20
HEPT (m)
0 0
10
15
20
25
30
35
40
45
vitesse linaire (cm/min)
On peut accrotre l'efficacit d'une colonne en diminuant v. Cependant en-dessous de 3 ml.min-1 HEPT passe par un minimum puis croit en raison de la diffusion longitudinale. V.1.3.2. Influence du diamtre des particules sur HEPT La rsistance au transfert de masse constitue le facteur limitatif la cintique des changes et pour augmenter celle-ci il faut diminuer au maximum la distance que doit parcourir le solut entre les diffrents sites, ce qui est possible en diminuant la taille des particules. Empiriquement il a t dmontr que
HEPT = B . dp
dp diamtre des particules; B et b constantes pour une colonne donne. Les valeurs de sont sensiblement constantes et voisines de 1,6 1,8.
HEPT (mm)
5 2 1 0,5 0,1 0
10
20
diamtre des particules (m)
La figure ci-dessus illustre le gain d'efficacit quand on passe d'une silice avec dp = 20 mm dp = 3 mm. Le remplissage des colonnes avec des phases stationnaires de fine granulomtrie entrane une
r Jean-Louis CUQ -
page 17
augmentation importante de l'efficacit, donc de la rsolution . Un compromis doit cependant tre ralis car si dp diminue, la perte de charge augmente de faon inversement proportionnelle dp2 P =
. L . v . 1 8 0 . 1 -
2 2
dp .
et des difficults de remplissage apparaissent. Lexemple de sparation chromatographique de phnothiazines obtenu pour deux dp est donn ci-dessous.
1 1
A
2 3
10
t R (min)
10
(min)
currentpoint
S N (CH 2)3 CH 3 N H CH3 Cl
S N (CH 2 )3 N (CH 2)3 N CH 3 CH 3 CH 3 Cl
S N (CH 2 )3 N CH 3 Cl
currentpoint
driv 1
driv 2
driv 3
Figure 4. Comparaison des chromatogrammes de 3 phnothiazines (k'1 = 3 ; k'2 = 6,5 ; k'3 = 15) en fonction du diamtre des particules d'un gel de silice. Colonne de 25 cm L et 2,1 mm de , v = 0,7 cm.s-1, dtection 254 nm. Phase mobile : actate d'thyle / mthanol / thylamine 33 % (80/20/0,25). A dp = 40 mm, P = 3 bars B dp = 10 mm, P = 35 bars
V.1.3.3. Influence des caractristiques de la colonne a) Influence de la longueur de la colonne Dans des conditions idales de remplissage de la colonne, la HEPT est indpendante de la longueur de la colonne et l'efficacit de la colonne est proportionnelle sa longueur HEPT = L / N. En ralit, selon dp, le remplissage de la colonne n'est pas idal et HEPT varie (Tableau 2 pour la phnothiazine 1). Rappelons que la perte de charge est proportionnelle la longueur de la colonne.
r Jean-Louis CUQ -
page 18
tableau 2. Variation de la HEPT pour la phnothiazine I en fonction de la longueur de la colonne (L) et de dp et en fonction du diamtre de la colonne. diamtre de la colonne 21 mm L (cm) HEPT (mm)
silice dp 10 m silice dp 10 m
colonne L = 15 cm
silice dp 10 m diamtre de la colonne (mm)
HEPT
(mm)
15 25 50 100
0,33 0,34 0,5 0,6 3,3
4,1 4,0 3,4 4,5
6,36 4,3 2,1
0,17 0,17 0,33
Le modle de colonne le plus utilis a 25 cm de longueur et 4,6 mm de diamtre. Ces colonnes sont en acier inoxydable. Remplies dune phase stationnaire avec des particules sphriques de 5 m de diamtre, elles permettent, selon la nature de la phase stationnaire, de raliser la plupart des sparations attendues dans les sciences des aliments. Des colonnes de 3 cm x 4,6 mm permettent des analyses de mlanges simples en routine. Dans le plupart des analyses une pr-colonne qui ne diffre de la colonne analytique que par sa trs faible longueur (par exemple 1 2 cm pour une colonne de 25 cm) permet de protger la colonne et den augmenter la dure de fonctionnement. b) influence du diamtre D'une faon gnrale les colonnes de faible diamtre (< 5 mm) sont plus efficaces, la diffrence tant d'autant plus grande que dp est petit (tableau 2). Le phnomne est d un remplissage plus homogne des colonnes de faible diamtre par rapport celui des colonnes de trs faible diamtre (cf IV-7). Les colonnes Microbore ont des diamtres internes de 1 2,1 mm. Leur mauvais succs rsulte surtout de la gnralisation dquipement adapts aux colonnes de 4,6 mm de diamtre. Le passage dune colonne de 4,6 mm 1 mm se traduit par une diminution de dbit dun facteur 21. Il faut alors disposer de pompes capables de dlivrer avec constance des dbits de lordre de 30 50 L.min-1. Si un systme dlution par gradient est utilis il faut alors disposer de pompes capables de dlivrer les phases mobiles avec des dbits contrls de quelques L.min-1.pour la pompe qui dlivre les plus petits dbits de phase mobile. Il existe des colonnes capillaires dont le diamtre varie entre 10 et 300 m. Elles sont remplies avec des particules de 0,2 0,5 m de diamtre. Par exemple une colonne de 192 mm x 0,3 mm permet la sparation de 5 protines en 30 secondes avec un dbit de 1,2 mL.min-1 ce qui reprsente une vitesse linaire de phase mobile de 128 cm.min-1. La pression atteinte nest que de 24 bar. De tels systmes qualifis de nano-chromatographiques. Dupont de Nemours propose de telles colonnes qui posent actuellement des problmes dutilisation du fait de la difficult trouver les pompes et les connexions ncessaires leur utilisation mais aussi en raison du manque de sensibilit de la plupart des dtecteurs. En effet les quantits danalytes sont trs faibles et souvent en dessous du seuil de sensibilit des appareils actuels.
r Jean-Louis CUQ -
page 19
V.1.3.4. Influence de l'injection L'injection en chromatographie en phase liquide est une opration dlicate actuellement automatise . a) Influence de la position de l'injection. La position de l'injection par rapport au niveau de la phase stationnaire revt une grande importance (figure 5). L'efficacit passe par un maximum quand l'injection est effectue quelques mm en-dessous du niveau suprieur de la phase stationnaire. b) Influence du volume inject. Le volume inject doit tre de quelques ml; s'il augmente, l'efficacit de la colonne diminue (figure 5).
disque poreux injection type I type II type III type IV 5 cm injection 8 mm cte 0 15 mm volume (l) type I II III IV I II III IV 36 83 37 58 43 95 45 64 HEPT (m)
1 silice dp = 5 m 10 4,8 mm
Figure 5. Influence du mode et du volume d'injection sur la HEPT (phnothiazine) v = 0,25 cm.s-1 De plus l'injection de grands volumes entrane souvent des dformations des pics. Il apparat des paulements ou des ddoublements qui indiquent alors une injection en deux temps. L'injection doit tre rapide et continue avec ou sans interruption de l'coulement de la phase liquide. En rsum, il est prfrable d'injecter 2 l d'une solution 10-3 M plutt que 20 l dune solution 10-4 M. c) Influence du type d'injecteur . Il existe deux types d'injection (figure 6) : - l'injection par seringue qui ne se rencontre pratiquement pas en HPLC. Il est impossible d'emprisonner le volume inject dans le disque poreux situ sur la tte de colonne, et de ce fait, il se produit une dilution importante du volume inject dans l'luant, ce qui entrane une efficacit plus faible qu'avec l'injection par seringue. - l'injection par vanne (boucle d'chantillonnage). Ce type d'injection qui est celui retenu dans les systmes automatiss, ne permet pas d'atteindre l'efficacit maximale.
r Jean-Louis CUQ -
page 20
disque d'tanchit
v
a
fritt colonne colonne
b
pompe colonne
b a
pompe
remplissage Injection par seringue
1/3 de tour
injection
Vanne boucle interne
Vanne boucle externe
pompe colonne boucle externe qualibre colonne
pompe boucle externe qualibre
remplissage
1/6 de tour
injection
Figure 6. Systmes d'injection de l'chantillon analyser en CPL
V.2. OPTIMISATION DU TEMPS D'ANALYSE Il est important, pour des raisons pratiques, que la dure de l'analyse soit la plus courte possible (environ 10 min). Pour une rsolution donne, il faut donc dterminer les conditions conduisant au temps d'analyse minimum. V.2.1. Optimisation du nombre de plateaux efficaces par seconde A rsolution et slectivit donnes, le nombre de plateaux efficaces ncessaires la sparation est fix et donn par :
Rs =
Dans ce cas, le temps d'analyse sera d'autant plus court que le nombre de plateaux efficaces par seconde sera plus grand.
1 - 1 . . 4
N ef
r Jean-Louis CUQ -
page 21
2 N ef = v . k' tr HEPT 1 + k'
a) Effet du terme en k' (figure 7)
k'
2
(1 + k' ) 3 0,15
0,10
0,05
10
15
k'
Figure 7. Variation de Les variations de
k'2 en fonction de k 3 (1+k)
k'2 en fonction de k' montrent que la courbe passe par un 3 (1+k') maximum pour k' = 2 si HEPT est indpendant de k'. b) Effet du terme v / HEPT
Les deux variables ne sont pas indpendantes HEPT = Avn et HEPT = B dp En combinant les deux relations on peut crire : HEPT = KH . dp . vn avec ~ 1,8 n = v . v n-1 v 0,3 < n < 0,7 1-n n v v 1 = v = HEPT HEPT K H. d2 K H . d2 p do p On peut augmenter ce rapport : - en augmentant v (puisque n < 1) - en diminuant le diamtre des particules. La perte de charge va cependant augmenterdans ces conditions. V.2.2. Pression et temps d'analyse * tR = L (1+ k') v HEPT = L N
tR = N (1 + k') HEPT avec ** HEPT = KH dp vn v
r Jean-Louis CUQ -
page 22
La perte de charge est donne par la loi de DARCY d2 Lv p 3 P = K = . K 180 (1 - )2 180 (1 - )2 P = J v.l. . avec J = d2 3 p En combinant *, ** et *** on obtient : a c K H . ( .J )b . N a . dp tR = (1 + k') Pb
a= 2 1 + n b= 1 - n 1 + n c= -
1 - n 1 + n
2 -
en prenant n = 0,5 et b = 1,8 KH1,33. (.J)0,33 . N 1,33 . dp1,73 ________________________________ P0,33
tR =
( 1 + k' )
A KH, J, N et k' constants, on peut donc diminuer tR en diminuant dp ou en augmentant P. La figure 8 montrent les variations de tR en fonction de P pour divers dp. Ainsi pour une pression de 70 atm, le passage de dp 40 5 mm permet de rduire le temps d'analyse de 7h30 10 min.
t R 5 10 h 10 4 1h d = 40 m (sec)
10
p d d d
10
30 min 10 min
p p p
= 20 m = 10 m =5 m pression (bars)
10
5 min 1 min 0 10 100 1000
10000
Figure 8. Variation du temps d'analyse en fonction de la pression diffrentes granulomtries = 1,8 cp ; = 1,05 ; Rs = 1 ; N = 28 250 ; n = 0,6. V.3. OPTIMISATION DE LA PERTE DE CHARGE Il est important de rduire les pertes de charge car travailler sous haute pression est toujours synonyme de difficults techniques. Pour maintenir RS et tR constants il faut que : 1 - le volume de la phase stationnaire reste constant
r Jean-Louis CUQ -
page 23
2 - le nombre total de plateaux thoriques reste constant quels que soient le dbit ou la longueur de la colonne 3 - le nombre de plateaux efficaces par unit de temps soit constant. HEPT = KH.dp vn
N=
= 1,8
et
tR=
n compris entre 0,3 et 0,7
L . 1 + k' v
L = const ant e HEPT HEPT v
do
t R = N ( 1 + k' ) .
k' est constant pour un compos donn avec phases stationnaire et mobile donnes. Il faut donc que L / v = constante Si on divise dp par 2, HEPT est divis par 2. Pour que tR reste constant il faut diviser L et v par 2 ce qui entrane que HEPT est divis par 2n. Il faut donc nouveau divisier L et v par 2 n et ainsi de suite. On obtient une srie convergente dont la somme des termes est :
21-n
Ainsi avec = 1,8 et n = 0,5 diviser dp par 2 permet de diviser L et v par 23.6 soit environ 12. Si dp est divis par 2 Lv . 180 (1-)2 P = _______________
dp .
2
P est divis par
23,6 . 23,6 ________ 22
soit par 37
En divisant par 2, on obtient une sparation identique quant sa dure et sa rsolution avec une colonne 12 fois plus courte et un dbit 12 fois plus faible ce qui ncessite une P 37 fois plus faible. Plus la colonne sera courte et plus son diamtre sera grand car Vs = Cte. Il faut signaler deux limites ces diminutions de L, v et dp 1 - il est impossible d'augmenter beaucoup le diamtre des colonnes sans entraner des perturbations notables dans l'coulement de la phase mobile. 2 - il est peu probable que l'on puisse diminuer le diamtre des particules en dessous de 1 m. V.4 . CONCLUSION L'optimisation d'une analyse par CPL est difficile en raison de multiples paramtres sur lesquels on peut agir. En fait, dans la pratique, on s'efforcera d'abord de choisir les conditions chimiques de la sparation (nature et composition chimique de la phase stationnaire, nature et composition chimique de l'luant, modification ventuelle des produits sparer) pour que le facteur de slectivit ne soit pas trop proche de 1 et pour les facteurs de capacit soient compris entre 1 et 10.
r Jean-Louis CUQ -
page 24
Le choix de la colonne (limit par les fournisseurs) se limitera celles de faible longueur (25 cm) et de 4,8 mm de diamtre par exemple. Les particules devront avoir un dp de l'ordre de 5-10 m. On veillera ce que l'injection soit la "moins mauvaise" possible. V.5. DETERMINATION GRAPHIQUE DES PARAMETRESopt Les principaux paramtres pour optimiser une analyse sont indiqus sur la figure 9 pour une viscosit de la phase mobile faible et gale 0,44 cP. Il existe de tels diagrammes pour des phases mobiles de viscosits autres.
L ( cm) 1000 300 100 30 10 100000 1000000 1000 P ( bar) 300 100 30 10 3
= 0,44 cp
10000
1000 1 3 20 30 100
( m)
30 10
t R (sec) 10000 3000 1000 300 100
10
0,5
0,1
v ( cm. sec
-1 )
5 (sec -1 )
10
20
50
100
200
HEPT ( m) 0,2 Nef / t
5000
500
100
20
figure 9. Dtermination graphique des paramtres optima en chromatographie en phase liquide
r Jean-Louis CUQ -
page 25
VI. DIFFERENTS TYPES DE CHROMATOGRAPHIE EN FONCTION DES PHENOMENES MIS EN JEU En chromatographie en phase liquide les principales interactions qui rgissent les mcanismes de rtention et dlution de soluts sont, en fonction de lnergie mise en jeu, lies des liaisons chimiques: - soit de Van der Waals (1 9 kJ.mole-1). Il sagit dinteractions dipolaires lies la prsence de diples induits et instantans ou permanents dans la molcules. Ces interactions existent dans pratiquement tous les systmes chromatographiques. Leur rle est souvent nglig en raison de leur faible nergie. Ces interactions se produisent faible distance (0,3 0,6 nm) et diminuent dintensit quand la temprature augmente. - soit hydrophobes (4 12 kJ.mole-1). Dans ce cas, cest lattraction entre deux (ou plus) molcules deau spares par une molcule ne donnant que peu ou dinteraction avec elles qui chasse cette molcule currentpoint
H O H
molcule apolaire
O H H
currentpoint Dans ce cas, moins la molcule est polaire (incapable de donner des interactions avec leau) et plus forte est la rpulsion. La chaleur augmente lagitation des molcules deau et augmente donc la force de rpulsion. Ces interactions se produisent entre 0,2 et 0,4 nm. - soit hydrogne (8 40 kJ.mole-1). Il sagit dune interaction dans laquelle deux atomes lectrongatifs dont lun est li un atome dhydrogne partagent ingalement cet atome. La distance de ces liaisons est denviron 0,2 nm. currentpoint
N H O C O H O
currentpoint
Ces interactions sont donnes par leau ; elles diminuent dintensit avec la temprature. - soit polaires ionises (lectrostatiques ou de Coulomb, 40 85 kJ.mole-1). Ces interactions entre ions dpendent, pour la plupart des groupes ionisables organiques, du pH. Ainsi par exemple les groupements carboxyles ne sont ioniss significativement qu des pH > pKa. Lintensit de ces liaisons de forte nergie diminue avec la temprature. - soit covalentes (330 400 kJ.mole-1). Lnergie de ces liaisons est trs lev et leur rversibilit ncessaire leur implication en chromatographie en phase liquide nest pas toujours accessible. Les seules liaisons covalentes rversibles utilisables en chromatographie en phase liquide sont les liaisons par pont disulfure. Ces liaisons sont peu sensibles aux variations de temprature dans des conditions de pH ou de potentiel doxydorduction donnes. Les diffrents systmes chromatographiques diffreront les uns des autres par la nature des interactions chimiques changes entre solut, solvant et phase stationnaire. Pour quune phase stationnaire soit utilisable il est donc ncessaire quelle donne une (ou plusieurs) interaction avec le solut (pour le retenir), mais il est aussi ncessaire que le solvant constituant la phase mobile donne des interactions avec le solut (pour lluer) et avec la phase stationnaire.
r Jean-Louis CUQ -
page 26
Il nexiste pas de phase stationnaire inerte et il faudra toujours se proccuper des interactions quelle est susceptible de donner mme dans des chromatographies o elle nintervient en principe que par un aspect strique .
VI.1. LA CHROMATOGRAPHIE D'ADSORPTION La phase stationaire est un adsorbant et la sparation est fonde sur les diffrences d'adsorption des molcules du mlange sur cette phase (CLS). Les premires sparations chromatographiques ont utilises comme phases stationnaires principales soit la silice, soit la cellulose. Il en rsulte que tous les modles raliss partir de ces phases reposaient sur un change analyte - phase mobile - phase stationnaire essentiellement du type liaison hydrogne. Sans indication quant la nature de la phase stationnaire, toutes les donnes qui seront inhrentes la chromatographie dadsorption seront donc fondes sur ce type dchange. C'est cette technique, couramment utilise qui offre les possibilits les plus vastes. Elle s'applique trs bien aux composs organiques dont la masse molaire est comprise entre 200 et 1000, que ces composs soient volatils ou non. Son domaine d'lection est la sparation de composs organiques renfermant des groupements fonctionnels diffrents ainsi que celle de certains types d'isomres. Elle s'applique mal la sparation de soluts trs peu polaires (par exemple ceux qui ne se diffrencient que par la longueur de leur chane aliphatique). VI.1.1. Thermodynamique de l'adsorption SNYDER (Principles of Adsorption Chromatography, M. Dekker, NY, 1968) a trait ce sujet en dtail. Le volume de rtention d'un solut est directement li la comptition entre les molcules de solvant M et de solut S la surface de l'adsorbant a. On admet gnralement que cette surface est recouverte d'une couche monomolculaire constitue par des molcules de la phase mobile M et de solut S. L'quilibre d'adsorption et de dsorption peut s'crire : currentpoint S (M ) + nM(a) currentpoint S(a) solut dans phase mobile phase mobile adsorbe solut adsorb
+nM(M) phase mobile "libre"
L'adsorption d'une molcule de solut S(M) currentpoint currentpoint S(a) se traduit par le dpart de n molcules de solvants de la surface de l'adsorbant nM(a) currentpoint nM(M). L'nergie d'adsorption est donne par : Ea = ES(a) + n EM(M) - ES(M) - n EM(a) Les termes des nergies en solution sont faibles et on a : Ea = ES(a) - n EM(a) ES(a) est constante et ne dpend pas du solvant. EM(a) dpend du solvant et du support mais pas de l'chantillon .
currentpoint
r Jean-Louis CUQ -
page 27
Une quation reliant le facteur de capacit k' du solut aux proprits de l'adsorbant a t propose par SNYDER VS o o log k' = log V a + (S - . A S ) + log VM Va : : S - AS S : As VS: VM : volume de phase mobile adsorbe par gramme d'adsorbant activit de l'adsorbant (en gnral comprise entre 0,5 et 0,9) est li aux proprits de l'luant et du solut. nergie libre d'adsorption du solut par unit de surface d'adsorbant dans des conditions d'activit standard (b = 1) : surface occupe sur l'adsorbant par une mole de solut : nergie libre d'adsorption des molcules de la phase mobile par unit de surface d'adsorbant volume phase stationnaire volume phase mobile.
G S = 2 , 3 RT
o o
G : variation d'nergie libre d'adsorption R : constante des gaz parfaits T : temprature absolue (Kelvin) Cette loi peut se simplifier de la faon suivante:
log k' = A -
.B
A et B = constantes
Il apparat alors que plus la valeur de o augmente et plus le log de k diminue et inversement. Il sagit donc l dun paramtre trs important dans ce type de chromatographie : il permet de matriser tR SNYDER a dcrit une srie luotropique (force luante d'un solvant) partir des valeurs de l'nergie libre d'adsorption e des molcules de solvant sur la surface de l'adsorbant (tableau 3). Tableau 3. Srie luotropique pour l'alumine. Pour la silice, les valeurs sont multiplier par 0,77. o n-pentane isooctane n-heptane cyclohexane cyclopentane sulfure de carbone CCl4 chlorure d'amyle ther isopropylique tolune chlorobenzne O 0,01 0,01 0,04 0,05 0,15 0,18 0,26 0,28 0,29 0,30 benzne bromure d'thyle ther thylique chloroforme chlorure de mthylne ttrahydrofurane mthyl thylctone actone dioxane actate d'thyle alcool amylique o 0,32 0,37 0,38 0,40 0,42 0,45 0,51 0,56 0,56 0,58 0,69 dimthylsulfoxyde aniline nitromthane actonitrile pyridine alcool isopropylique alcool thylique alcool mthylique thylne glycol acide actique o 0,62 0,62 0,64 0,65 0,71 0,82 0,88 0,95 1,11 grand
r Jean-Louis CUQ -
page 28
Si o augmente, log k diminue et la rtention diminue (avec ce type de chromatographie dadsorption phase stationnaire polaire) VI.1.2. Adsorbants utiliss en CLS Le processus de l'adsorption est complexe et les forces mises en jeu qui dpendent du solut, du solvant et de l'adsorbant sont nombreuses (forces de dispersion de London, forces lectrostatiques, forces de liaisons hydrogne etc...). Il devient alors impossible de prciser la part relative de ces diffrentes forces d'adsorption car pratiquement on n'observe que leur rsultante. Par ailleurs, du fait de la prsence d'eau (mme l'tat de traces) dans les phases stationnaire et mobile il est galement impossible de prciser la part de la chromatographie de partage ct de la chromatographie d'adsorption. Malgr ces nombreuses interactions, les prvisions en chromatographie liquide d'adsorption sont relativement simples (particulirement quand des adsorbants polaires o les liaisons hydrogne constituent les forces dominantes sont utiliss : silice ou alumine ou cellulose). SNYDER distingue ainsi 4 types d'adsorbants I II III IV inorganiques polaires inorganiques apolaires greffs polaires greffs apolaires silice, alumine graphite, charbon aminopropyl (NH2), cyanopropyl (CN) C18 - C8
VI.1.2.1. Adsorbant de type I - Silice ou Alumine Les sites actifs sur lesquels s'adsorbent les molcules de soluts sont, pour la silice, des groupes silanol*. Ces groupes sont de trois types : H O Si ssilanol isols H O Si H O Si O Si H O Si H HO Si silanols gminaux OH
silanols liaison hydrogne - H2 O O Si siloxane Si liaison hydrogne
isol
Pour des pH suprieurs 8 la silice nest pas stable et se dissout. Toutes les phases qui utiliseront une base silice seront donc limites au niveau du pH de leur phase mobile 8. En prsence d'eau, la plupart de ces groupes sont associs cette molcule. Cette eau doit tre limine par chaufface prolong 150C. Un chauffage temprature suprieure 200C se traduit partir des silanols liaison hydrogne, par la formation de siloxane dont le caractre apolaire est important.
r Jean-Louis CUQ -
page 29
OH
O Si
O O Si O
O
O
OH
OH
Si O
O
Si
OH
O Si O
OH
Si O
Si O O O Si O O O
Si O O Si
OH
OH
+ :NR3
Si O O Si
OH
:NR3
Si
Si
OH
L'exprience montre que la silice convient bien la sparation de soluts qui sont des bases au sens de Brnsted (exemple : amines qui se fixent nanmoins de faon souvent de faon irrversible) ; on dit que cet adsorbant a un caractre acide. Le gel de silice est prpar par prcipitation acide de silicate de sodium : il se produit une polymrisation de lacide silicique form en un rseau appel hydrogel. En solution dilue un gel faible ou un prcipit est obtenu tandis quen milieu concentr lhydrogel est ferme. Le schage de ce dernier conduit un xrogel (photographie b obtenue par microscopie lectronique balayage).
Les gels de silice prsentent une structure poreuse. KIRKLAND J.J. (LC.GC, 1996, 14, (6), 486-500) a obtenu la glification de silicates solubles par cohalescence avec formation de structures de type sil-gel (photographie a obtenue par microscopie lectronique balayage). Les subparticules de ce gel sont homognes; la porosit est de 50%; le gel a une surface de 150 m2 / g et les pores ont 10 nm de diamtre. Relativement stables aux pH levs. Le xrogel b est spongieux; sa porosit est de 70% et sa surface de 300 m2 / g. Les pores ont un diamtre de 10 nm Quand la surface est compltement hydroxyle, le gel de silice a une densit de groupements silanols de 8 mole / m2. Ces silanols sont localiss pour la plupart dans des micropores de 1 nm de diamtre ce qui ne les rend accessibles qu des analytes de faibles dimensions molculaires.
La silice peut tre utilise en milieux aqueux et non aqueux.
r Jean-Louis CUQ -
page 30
Inversement, l'alumine convient bien pour la sparation de soluts acides (phnols, acides carboxyliques) ; cet adsorbant est dit basique.
Al O O Al O Al Al O OH OH OO O OH H OR
Al
O Al O
C O
O Al O Al
Al
O Al O
a) Influence des groupements fonctionnels du solut
Avec ces adsorbants de type I, les molcules polaires sont toujours plus adsorbes que les molcules non polaires. amides > amines > acides carboxyliques >alcools > ctones > esters > composs nitrs > thers > hydrocarbures aromatiques > hydrocarbures non saturs > hydrocarbures saturs.
b) Influence de la force d'lution du solvant
La force lutive d'un solvant est caractrise par (tableau 3). Une augmentation de 0,05 de la force lutive diminue environ de moiti la valeur du facteur de capacit k'. En pratique, on recherche une valeur de force lutive qui donne une rtention convenable.
c) Diffrents types de support
Deux paramtres caractrisent ce type de phase stationnaire : - la granulomtrie. Ainsi de 60 m pour dp pour la chromatographie liquide/solide classique, on est pass des valeurs de dp comprises entre 1 et 10 m. - la forme. Les grains de silice avaient l'origine une structure irrgulire. Actuellement ces grains sont le plus souvent sphriques. Il existe des grains constitus par un noyau compact ( 30 m) recouverts d'une fine pellicule poreuse (1 m) : ce sont les supports pelliculaires.
Gnralits sur les phases greffes sur les particules de silice.
Ces phases reposent sur ltablissement dune liaison silylther. Elle est obtenue par raction de la silice avec des drivs du type monochlorodimthylsilane ou trichlorosilane pour les plus importants.
CH 3 Si O H + Cl Si CH 3 monochlorodimthylsilane R CH 3 Si O Si CH 3 R
Si
O H + Cl
Cl Si Cl R
Si
O Si
Cl R
Si
O H
Si
trichlorosilane
r Jean-Louis CUQ -
page 31
La liaison Si - O - Si nest pas stable pour des pH infrieurs 2. La plage dutilisation des phases greffes stend donc de pH 2 pH 8. Nanmoins de nombreux travaux ont t raliss pour largir cette gamme. Par exemple la substitution des groupements mthyls par des groupes plus volumineux (propyls par exemple) minimise les possibilits dhydrolyse et permet donc dlargir la gamme dutilisation possible de ces phases stationnaires.
CH3 CH3 Si O Si CH3 Si Si OH CH3 O Si CH3 Si OH Si Si Si Si O CH2 CH2 Si
CH2 CH 2 CH3 CH2 CH2 O Si CH3 CH2 CH2 OH
silice greffe avec du chlorodimthyloctadcyl silane
silice greffe avec du chlorodipropyloctadcylsilane
IV.1.2.2. Adsorbant de type III. Phases greffes polaires Les principaux groupes fonctionnels des phases greffes polaires sont indiqus dans le tableau 4. Ces phases sont classes en fonction de leur polarit.
analyse de :
- faiblement polaires (glycophase ou diolphase dimthylamino) - moyennement polaires - fortement polaires (cyano) (amino, diamino)
composs carbonyls phnols , amines , esters oses, osides, peptides, nuclotides
Un exemple de chromatrogramme doses obtenu sur une silice greffe NH2 est donn sur la figure 10.
2 1 4 3 5
t 0 10 20 R
(min)
Figure 10. Sparation fructose (1), glucose (2), saccharose (3), maltose (4) et lactose (5) sur Si greffe NH2. dp = 10 mm. Colonne L = 30 cm, = 3,9 mm. Eluant actonitrile/eau (80/20). Dbit 2 ml/min. Dtection rfractomtrique.
r Jean-Louis CUQ -
page 32
Tableau 4. Type de phase
Groupes fonctionnels de phases polaires greffes et applications principales Structure du greffon CH2 OH Si O Si (CH2 )3 O CH2 CH OH Applications quinones, flavones, amines, peptides, protines,etc... glucides (oses), vitamines, isomres D-L, peptides, alcools, nuclosides, etc... cf diol CH3 NH 2 cf amino lipides, strodes, pesticides, phnols, amines, esters, etc..
glycophase (Diol)
amino
Si O Si Si O Si Si O Si Si O Si
(CH2 )3 NH 2 CH3 (CH2 )3 N (CH2 )3 (CH2 )2
dimthylamino diamino cyano
NH (CH2 )2 C N
VI.1.2.3. Adsorbant de type IV. Phases greffes apolaires Les phases greffes apolaires reprsentent les phases stationnaires les plus utilises en HPLC. Ces phases (qualifies de rverses ou d'inverses) sont prpares partir de silice par formation dune liaison thersilyl (cf gnralits sur les phases greffes) La longueur du greffon alkyle R sil sgit dune chane aliphatique ou la nature mme de cette chane (groupements phnyl, etc) sert caractriser la phase.
RP 8 implique R = (CH2)7 - CH3 (octyl) RP18 implique R = (CH2)17 - CH3 (octadcyl) . Les phases RP 8 et RP 18 sont les plus rpandues et les plus utilises.
Les applications possibles de ce type de phases stationnaires sont trs nombreuses: RP 8 substances lipophiles ou moyennement polaires Bien adaptable la chromatographie par appariemment d'ions Substances polyaromatiques, antibiotiques, vitamines hydrosolubles, acides et esters organiques, hydrocarbures, corticostrodes, barbituriques, phospholipides, etc... Substances lipophiles, acides amins, etc..
RP 18
En ralisant une surface hydrophobe on inverse totalement la nature des interactions fournies par la silice. L'eau n'a pas d'affinit pour le greffon hydrophobe et la force luante des solvants est exactement l'inverse de celle qui est observe sur supports polaires d'o le terme reversed phase (RP) appliqu ce genre de support.
Les soluts les plus polaires seront les moins retenus, les soluts apolaires seront retenus dautant plus fortement que leur hydrophobicit est leve.
r Jean-Louis CUQ -
page 33
La rtention des composs apolaires croit avec la longueur du greffon et avec le taux de greffage de la surface (surface coverage). La taille des particules n'a pas d'effet sur la rtention exprime par le facteur de capacit k', les effets de la taille ne concernant que la pression et la HEPT. a) mcanisme de rtention Malgr tous les travaux qui lui sont consacrs, il existe encore beaucoup de flou dans son interprtation. Dans la thorie solvophobique, ce sont les interactions apolaires dans un solvant polaire qui sont dcrites. On peut schmatiser les forces mises en jeu, forces dont la rsultante est la force de liaison entre le solut et la phase greffe (figure 11).
zone hydrophile eau particule de silice greffon apolaire 1 2 3 1 forces de Van der Waals entre solut et phase stationnaire 2 forces lies l'hydrophobicit 3 interactions hydrophobes entre molcules de soluts 4 interactions polaires entre molcules de soluts et entre molcules de soluts et l'eau
zone hydrophobe du solut
Figure 11. Reprsentation schmatique des interactions mises en jeu dans la chromatographie en phase inverse. Le coefficient de distribution (ou coefficient de partage) est : K = CS / CM La variation d'nergie libre DG de la liaison du solut la phase greffe est gale : G = - RT Ln K Le facteur de capacit k' est gal : k' = K .VS / VM o VS G ln k' = ln VM RT Avec VS volume du solut dans la phase stationnaire et VM volume du solut dans la phase mobile. Pour un luant donn et une phase stationnaire greffe donne, on constate que le facteur de capacit k' augmente avec l'hydrophobicit du solut. Ainsi les variations de log k' en fonction du nombre de groupements mthylne (CH2) dune famille de composs organiques donns sont linaires (figure 12, a ).
log k'
2 1 0 -1
d c b a
ln k'
4 2 0 -2
acides acides amins
amines
n CH
60
100
140 surface molculaire o apolaire (A )2
figure12. Variations du logarithme du facteur de capacit en fonction
a. du nombre de CH2 d'alkyl benznes (n = 0 = benzne). Phase stationnaire greffe en C8. mthanol / eau : a = 1/0, b = 9/1, c = 8/2, d = 6 / 4 b. de la surface molculaire apolaire
r Jean-Louis CUQ -
page 34
Pour des composs biologiques (amines, acides carboxyliques, acides amins), on observe galement une relation linaire entre ln k' et la surface apolaire de la molcule (figure 12,b). Plus l'ionisation augmente et plus le facteur de capacit diminue. Pour les acides monoprotons, le facteur de capacit est gal : Ka k' 0 + k' -1 + + H H COO k' = avec K a = COOH Ka 1 + + H
currentpoint pour lquilibre COOH currentpoint + H+ COOk'o facteur de capacit de la forme non protone (COOH), k'-1 facteur de capacit pour la forme dprotone (COO-), Ka constante de dissociation de l'acide et H+concentration en proton. Le facteur de capacit sera donc le plus lev pour ces acides carboxyliques quand les conditions de pH empcheront lionisation de la fonction acide cest dire pour des valeurs de pH infrieures au pKa de lacide. Gnralement on ajoute la phase mobile un acide fort dilu (acide phosphorique par exemple) pour atteindre ces valeurs de pH qui doivent nanmoins rester suprieures 2 pour viter lhydrolyse de la liaisons silylther. Pour les bases faibles, on a une expression similaire (amine par ex.). Il faut remarquer ici que les valeurs des pKa de ces bases sont souvent suprieurs la limite de stabilit pH des silices. De fait il nest pas dexemple danalyse des pH suprieur 12. currentpoint NH3+ currentpoint+ NH2 + H
k' 0 + k' 1 k' = 1 +
(K ) H
a + a +
(K ) H
avec K a =
NH 2
+
NH 3
avec Ka constante de dissociation de la forme protone, k'1 facteur de capacit pour la forme protone (NH3+). Les variations de k' pour quelques acides carboxyliques sont indiques sur la figure 13.
k'
15 3 10 2 1 5 4
OH OH OCH 3 OH CH2 COOH a.homovanillique
currentpoint
COOH acide benzoque
COOH OH CH 2 COOH
a.salicylique
pH 2 4 6
a.3,4-dihydroxyphnylactique
Figure 13. Variations du facteur de capacit pour quelques acides mono protons en fonction du pH (chromatographie en phase inverse C8) 1 acide benzoque; 2 acide homovanillique; 3 acide salicylique; 4 acide 3,4 - dihydroxyphnylactique.
currentpoint
r Jean-Louis CUQ -
page 35
L'effet de la temprature sur le facteur de capacit peut tre tudi partir de la loi de VAN'T HOFF (effet de la temprature sur la constante d'quilibre) o o d ln K H H soit ln K = + cte = dt 2 RT RT Ici on aura: o o VS H S ln k' = + + ln RT R VM
H variation d'enthalpie
et
S variation d'entropie.
Si on porte ln k' en fonction 1 / T, Ho correspondra la pente de la droite. Ce type de reprsentation est indiqu sur la figure 14 pour quelques acides aromatiques. On constate que quand la temprature augmente k' diminue.
ln k' 2 2,3 3 2 3 0 1 1
currentpoint
OH OH OH OH OH CH2 OH CH COOH COOH CH2 COOH
-2,3
2,9
3,1
3,2
3,3
3 10 / T
1 currentpo
Figure 15. Variations en ln k' en fonction de 103/T (Van't Hoff) pour quelques acides armatiques en chromatographie en phase inverse (C8). Eluant tampon phosphate 50 mM pH2 ( ), et mme tampon additionn de 5 % d'actonitrile (1, 2, 3) 1 = acide dihydroxymandlique 2 = acide hydroxyphnylactique 3 = acide dihydroxyphnylactique. b) influence de la phase mobile L'eau constitue la base de toute phase mobile en chromatographie liquide sur phases greffes apolaires.
Elution en mode isocratique
Dans ce type dlution, la composition de la phase mobile reste constante tout au long de lanalyse chromatographique. On utilise gnralement soit un mlange binaire type eau / mthanol, eau / actonitrile ou eau / ttrahydrofurane, soit un mlange ternaire eau / actonitrile / mthanol. Ce type dlution est applicable des analytes dont les valeurs de k ne sont pas trop diffrentes. On constate gnralement une variation linaire du log k' en fonction du pourcentage d'eau dans la phase mobile (figure 15) (voir aussi figure 12).
r Jean-Louis CUQ -
page 36
log k' 1
nitrobenzne
tolune 0 phnol aniline -1 % d'eau
20
40
Figure 15. Variations du log k' en fonction de la teneur en eau du mlange mthanol / eau en chromatographie en phase inverse. La phase stationnaire subit des changements de structure selon la nature de la phase mobile. Il nest pas actuellement possible de prvoir de tels changements et seule lexprimentation permet den dterminer les effets. Ainsi l'ordre d'lution de composs benzniques varie avec l'luant (Tableau 5 et figure 16). Tableau 5. Ordre d'lution de drivs benzniques en fonction de la nature de l'luant. Chromatographie en phase inverse (C18). MeOH/eau 50/50 - CONH2 - CH2OH - OH - CHO - CH2-CH2 OH - CN - COCH3 - NO2 -H - OCH3 - COOCH3 - CH3 - Cl - COOCH2CH3 ACN/eau 30/70 - CONH2 - CH2OH - OH - CH2 -CH2OH - CHO - COCH3 - CN - NO2 - COOCH3 -H - OCH3 - COOCH2CH3 - CH3 - Cl THF/eau 25/75 - CONH2 - CH2OH - CH2-CH2OH - CHO - COCH3 - OH - CN - COOCH3 - NO2 - OCH3 -H - COOCH2CH3 - CH3 - Cl
r Jean-Louis CUQ -
page 37
absorbance 254 nm 1 1
currentpoint
O
1
0,5
NH 2
OCH 3
CH 2OH
OCH 3
CH 2 CH 2 OH
CH 2 CH 3
0 0 5 10 temps de rtention (min)
CH 2
OH
CH 3
4 5
NO 2
currentpoint
Figure 16. Chromatogramme en phase inverse C18 (dp = 10 m), colonne 250 x 4 mm. Solvant CH3CN/eau 49/5, (v/v). Dbit 1,2 ml.min-1, P 60 bars. Par ailleurs, SHOEMAKERS et al. (1978, J. Chromatography) ont montr que le facteur de capacit tait reli la concentration en solvant organique par la relation: ln k = A 2 + B + C avec fraction de solvant organique dans la phase mobile. Mais cest SNYDER et al. (1979, J. Chromatography) puis CSOKAN P. et al. (LC-GC, 1993, 6, n 6, 361-369) qui ont montr que la valeur du facteur de capacit dans une phase mobile compose deau et de solvant B tait gale :
log k = log keau + S .
avec k facteur de capacit, keau facteur de capacit avec 100 % deau, S constante pour chaque analyte avec des valeurs typiques de 4 pour les petites molcules (poids molculaire infrieur 1000) et le pourcentage de solvant B. Cette quation peut tre valide par deux ou trois chromatogrammes. L'interaction de Van der Waals varie avec la surface de contact entre la partie apolaire du solut et la phase stationnaire greffe. Ainsi, les phases greffes possdent une capacit de reconnaissance suivant la forme de la molcule. Il sera ainsi possible de sparer certains isomres de position, par contre la sparation des isomres gomtriques (cis/trans) ou optiques (D/L) est plus difficile (cf phases greffes chirales.)
currentpoint
CH3 CH3 CH CH 2 CH COO
leucine norleucine
CH3
CH 2 CH CH3 CH
COO NH3+
isoleucine
NH 3+ COO
CH3
(CH2)3
CH
+ NH 3
currentpoint
r Jean-Louis CUQ -
page 38
Les rtentions sont lies aux solubilits dans la phase mobile. Si l'on ajoute un sel neutre, l'activit de l'eau diminue et log k' = f ([sel]) est linairement dcroissant. En conclusion on peut dire que plus le solut sera soluble dans la phase mobile, moins il sera retenu. Elution en mode gradient En mode gradient, le facteur de capacit moyen k* est analogue au k en mode isocratique. Les mmes modifications du chromatogramme apparaissent pour k* comme pour k. Par exemple la rtention reste toujours plus grande avec des valeurs leves de ces deux paramtres. Le mode gradient permet llution de composs dont les k* sont trs diffrents (pente de variation du solvant leve) ou au contraire de composs dont les k* sont trs voisins (pente de variation nulle : isocratique). Si tous ces composs sont prsents dans lchantillon analyser, il est clair quil faudra faire varier dans le temps la concentration en solvant de faon diffrente, do la ncessit dutiliser un systme dlution gradient. Dans ce dernier cas, les meilleurs rsultats sont obtenus pour des valeurs de k* comprises entre 2 et 10. Il faut donc adopter une phase mobile dont la composition permettra dobtenir la valeur dsire de k*. En mode isocratique k nest pas affect par des variations de longueur, de diamtre, de dbit, et par la taille des particules, ce qui permet des changements dchelle relativement simples. En mode gradient, k* est modifi par de tels changements et le changement dchelle reste toujours dlicat. Cest SNYDER et al. (Practical HPLC Method development, 2nd Edition, Wiley Interscience, New York, USA, 1997) qui dcrit le plus en dtail les paramtres prendre en considration dans ces systmes dlution. Le facteur de capacit moyen en mode gradient est gal :
k* = t G . D . 100 VM . S . avec S = d(log k) d
avec tG le temps dans le systme gradient, D le dbit, VM le volume de phase mobile dans la colonne, S constante pour chaque analyte avec des valeurs typiques de 4 pour les petites molcules (poids molculaire infrieur 1000), DF est la modification de la fraction volumique de B (0 100). En mode gradient on a donc : k* . VM . S . tG = D . 100 Pour k* dsir de 5 et une valeur moyenne de S gale 4, cette quation devient :
tG =
20 . VM . D . 100
VM est gal VM = . VT . Avec voisin de 0,5 on a alors : 2 L . . dC . 2 VM = 0,5 . L .d C 4 (L longueur de la colonne, dC diamtre interne, porosit interstitielle, VM volume de phase mobile). Ainsi avec une colonne de 15 cm x 0,46 cm : VM est environ de 1,6 mL. Avec un gradient de 5 100 % de solvant B ( = 95) et D = 1,5 mL.min-1 tG est gal 20 min. Dans le systme considr, lanalyste obtiendra donc un chromatogramme satisfaisant en ralisant un gradient en 20 minutes, la fraction de B passant alors linairement de 5 100 % dans la phase mobile.
r Jean-Louis CUQ -
page 39
Les appareils modernes proposent des systmes grs par microordinateurs qui permettent de raliser des gradients par mlange de deux solvants. Le plus souvent les gradients raliss sont linaires, cest dire que les variations de B dans la phase mobile en fonction du temps sont linaires. Certaines appareils permettent des gradients curvilignes ou complexes et il existe mme des systmes qui autorisent la mise en place de gradients par mlange ternaire (3 solvants) voire quaternaire. Dans ces cas seule lexprimentation et le savoir faire de lanalyste peuvent rpondre un besoin analytique : les paramtres prendre en considration restent cependant trs nombreux et il devient souvent illusoire de matriser toutes les inconnues introduites dans ces conditions. c) Augmentation de k' par modification du solut a) fixation sur un solut polaire d'un compos apolaire par liaison covalente Cette mthode est de plus en plus largement utilise pour sparer les acides amins par chromatographie en phase greffe apolaire. Il existe aujourdhui de nombreuses ractions chimiques de modification/dtection des acides amins. La plupart de ces ractions ont les particularits suivantes: elles sont rapides, stoechiomtriques, et donnent des drivs dont certains sont trs stables et de dtection facile. La sensibilit de dtection a t multiplie par un facteur 1000 en comparaison avec la dtection ninhydrine, par lutilisation de produits donnant avec les acides amins des drivs fluorescents. 1) Raction avec l O-phtaldialdhyde (OPA) LOPA ragit froid, en prsence de mercaptothanol et en milieu alcalin avec les acides amins pour donner des drivs isoindoliques fluorescents ( ex = 340 nm , m = 455 nm ).
currentpoint
CHO CHO
HS CH CH2 OH 2 NH 2 CH COOH R
S CH2 CH2OH N CH COOH R + 2H 2O
currentpoint
La proline et lhydroxyproline ne donnant pas directement la raction, il est ncessaire, pour pouvoir les dtecter, de traiter au pralable les hydrolysats par de lhypochlorite de sodium. La stabilit de certains drivs isoindoliques forms nest pas trs grande, ce qui implique donc une analyse extemporane. 2) Raction avec la fluorescamine ( 4-phnylspiro ( furan-2(3H),1-phatal)-3,34 dione ). La raction se fait temprature ambiante et est complte entre pH 7 et 9 en environ 1 seconde ( ex = 390 nm et m = 475 nm ). currentpoint
COOH R O O O O NH2 CH R COOH CH N OH O COOH
+H2 O
currentpoint
Il sagit dune raction trs sensible (seuil de dtection voisin de la pmole) qui permet des dosages de quantits dacides amins de lordre de 0,1 nmole. La fluorescamine ne ragit pas non plus avec les amines secondaires comme la proline ou ses driv. Pour raliser le dosage de ces acides amins il faut donc les transformer en amines primaires par traitement lhypochlorite ou la N-chlorosuccinimide .
r Jean-Louis CUQ -
page 40
3) Raction avec le chlorure de dansyle Ce ractif permet dobtenir des drivs sulfamides trs fluorescent .
currentpoint
CH3 N CH3 NH2 CH COOH R CH3 N CH3 SO 2 NH CH COOH R + H Cl
+
SO2 Cl
currentpoint
Il existe actuellement beaucoup dautres mthodes de dtection fluorimtrique des acides amins (FMOC etc.). Il sagit toujours de mthodes trs performantes dont le choix est fonction de paramtres comme la stabilit des drivs forms, la simplicit de mise en oeuvre, le cot etc. Il faut signaler que tous les drivs dcrits (OPA, fluorescamine, dansyl ) absorbent dans lUV, ce qui permet leur dtection et leur dosage avec une sensibilit moindre que par la mthode fluorimtrique. 4) Raction au phnylisothiocyanate ( PITC ) Le phnyl isothiocyante ragit avec les acides amins pour former des drivs facilement dtectables dans lUV. Les phnylthiocarbamyl (PTC) sont transformables en drivs phnylthiohydantone (PTH ) en milieu chlorhydrique en prsence de mthylnitrosamine.
H Cl , CH 3 NO 2 NH C S NH CH R COOH N O S NH R
N C S
NH2
CH
OH
COOH
+H O 2
5) Raction avec le 9,fluornylmthyl-chloroformate (FMOC-HCl) currentpoint + NH 2 - R
CH2 O C Cl O
pH 8 , 60C 5 min
CH2 O C O NH R
currentpoint
6) Raction avec le chlorure de Dabsyl
currentpoint
CH3 N CH3 N N SO2Cl
NH 2 -R
pH 9 70C 10 min
CH3 N CH3 N N SO2 NH R
currentpoint
Les drivs obtenus ont des facteurs de capacit beaucoup plus importants que l'acide amin de dpart. Il est ainsi possible de raliser leur sparation par HPLC en phase inverse (figure 17). De plus ces drivs sont dtectables par fluorescence ou par absorption UV, ce qui facilite le dosage. La limite de dtection atteint respectivement 10-13 et 5.10-12 g. Les avantages et inconvnients des principales mthodes de drivation des acides amins sont indiques dans le Tableau de synthse ci-dessous
r Jean-Louis CUQ -
page 41
dabsyl amines 2aires oui sensibilit 5 pmoles UV stabilit bonne interfrence du ractif oui automatisable moyen
mode de drivation PITC OPA oui 5 pmoles UV moyenne oui mauvais
FMOC
non oui 150 fmoles Fluo 100fmoles Fluo mauvaise bonne non oui oui moyen
Il existe des possibilits de combinaisons de certaines de ces mthodes. Ainsi la combinaison OPA FMOC permet dobtenir aprs chromatographie en phase inverse et par mesure de labsorbance 338 nm tous les acides amins avec des NH2 primaires jusqu llution de la lysine puis, par lecture 262 nm llution et la dtection de la proline et de lOH-proline (GODEL H. et al, 1992, 5, LC-GC, 44-49 ).
14 8 1 2 3 4 7 6 9 11 10 12 15 13 18 16 17
A
5
10
20
temps de rtention (min)
6 4 1 23 22 2 3 8 5 9
12 10
13 19 14
20 11 16 15
17
21
temps de rtention (min) 5
10
Figure 17 : Chromatogramme en phase inverse (C18) des drivs dacides amins A ractif OPA - drivs isoindoliques ; dtecteur fluorimtrique B drivs PTH 1.Asp, 2.Glu, 3.Asn, 4.Ser, 5.Gln, 6.His, 7.Gly, 8.Thr, 9.Arg, 10.Ala, 11.acide -butyrique, 12.Tyr, 13.Met, 14.Val, 15.Phe, 16.Ile, 17.Leu, 18.Lys, 19.Pro, 20.Trp, 21.NorLeu, 22.acide cystique, 23.CH3Cys
r Jean-Louis CUQ -
page 42
b) Fixation sur un solut ionique d'un compos apolaire par liaison ionique : Appariement d'ions Le but de cette mthode est de lier, par formation dune paire dion, une molcule polaire ionise (acide, base par exemple) qui nest donc pas directement analysable en HPLC-RP, un ion de charge oppose mais porteur dun groupement apolaire qui permet lanalyse dans ces conditions. + analytite ractif formeur de paire d'ions + currentpoint (A+)M + (B-)M currentpoint (A+ B-) s A+M solut ionique (ion) B-M ou (B+) solut ionique apolaire (contre-ion) htarion +B-) paire d'ion (A + AB s K AB = + A M B M La paire d'ion forme peut possder un facteur de capacit autorisant une bonne sparation chromatographique sur phase stationnaire greffe apolaire. k' sera d'autant plus grand, que l'hydrophobicit du contre ion sera grande.Les principaux ractifs formeurs de"paire d'ions" sont indiqus dans le tableau 6.
Tableau 6. Principaux ractifs formeurs de paires d'ions (B+) (B-) bromure d'hexadcyltrimthyl-ammonium bromure de ttrabutylammonium 1 heptane sulfonate 1 hexane sulfonate (Na+) (Na+) (Na+) (Na+) (Na+)
bromure de ttradcyltrimthyl ammonium 1 octane sulfonate bromure de ttramthyl ammonium bromure de ttrapropyl ammonium 1 pentane sulfonate Dodcyl sulfate de sodium Naphtalne sulfonate
On peut par exemple obtenir des sparations d'acides organiques (figure 18) ou d'amines (figure 19).
2 1 3 4 5 6 7
10
20
(minutes)
1 = acide 4-aminobenzoque ; 2 = acide 3-aminobenzoque ; 3 = acide 4-hydroxybenzoque ; 4 = acide 3hydroxybenzoque ; 5 = acide benzne sulfonique ; 6 = acide benzoque ; 7 = acide tolune 4-sulfonique.
Figure 18. Chromatographie par appariement d'ions en phase inverse d'acides organiques (RP2) Phase mobile 0,03 M ttrabutylammonium pH 7,4.
r Jean-Louis CUQ -
page 43
DHMA VMA MGA
SH/AA
DOPAC
N.Syn
DOPA+NM
Isopron
3-H-tyrm
Syn
20
Tyrm
40
3-M-tyrm t (minutes) R
NE
Figure 19. Sparation isochratique de catcholamines par chromatographie par appariement d'ions en phase inverse. Le contre-ion est le SDS. L'ion d'appariemment (hetrion : ion compagnon) se trouve dans la phase aqueuse une concentration beaucoup plus leve que celle du solut A+. Le coefficient de distribution du solut est : [A+B-]s _______ = KAB [B-]M Ks =
[A+]M Le facteur de capacit est proportionnel Ks en chromatographie de phases inverses et 1/ KS en chromatographie dite normale, k' sera proportionnel la concentration de l'agent d'appariement dans le premier cas, et son inverse dans le second.
VI.2. LA CHROMATOGRAPHE DE PARTAGE
Dans cette technologie la phase stationnaire est un liquide qui imprgne un support en principe inerte ou est greffe par liaison chimique covalente sur ce support. La sparation des soluts est fonde sur leur partage entre cette phase stationnaire et la phase mobile liquide. Le coefficient de distribution est appel coefficient de partage : CS VS K= et k' = CM VM k' facteur de capacit VS volume de phase stationnaire contenue dans la colonne VM volume de phase mobile contenue dans la colonne CM Cs : concentrations respectives du solut dans la phase mobile et dans la phase stationnaire. Cette technique s'apparente l'extraction liquide/liquide base sur les diffrences de solubilits dans deux phases non miscibles, mais ici une des deux phases est immobilise sur un solide dont les particules ont des diamtres trs petits (dp) ; l'augmentation considrable de la surface de contact entre les deux phases fait que l'on obtient des efficacits trs suprieures celles obtenues en extraction liquide/liquide classique contre-courant.
MN
r Jean-Louis CUQ -
page 44
Comme en extraction, il faut que la solubilit de l'une des deux phases dans l'autre soit aussi faible que possible. Dans la chromatographie de partage classique, on choisit une phase stationnaire polaire et une phase mobile apolaire. Dans la chromatographie de partage polarit de phases inverse, on choisit une phase stationnaire apolaire et une phase mobile polaire. La chromatographie de partage donne de trs bons rsultats pour la sparation de composs groupements fonctionnels diffrents, de composs d'une srie homologue. La chromatographie liquide-liquide classique (phase stationnaire polaire) est surtout utilise pour la sparation de composs polaires, tandis que la chromatographie polarit de phase inverse est utilise pour la sparation de composs apolaires (hydrocarbures longue chane).
VI.2.1. Phases stationnaires utilises en chromatographie liquide / liquide
a) Tous les supports utiliss en chromatographie solide-liquide peuvent tre utiliss condition qu'ils soient rendus inertes. On utilise soit : des supports poreux tels que la silice sur laquelle la phase stationnaire liquide est une multicouche lie au support par des liaisons hydrogne ou polaire aux groupements silanols. Il existe des supports pelliculaires (poreux en surface). des supports poreux apolaires tels que la silice dans laquelle les groupements silanol ont t transforms en groupements trimthyl apolaires (ou hexamthyl) par raction avec le trimthylchlorosilane. Il s'agit ici d'une chromatographie en phase inverse. currentpoint
CH 3 Si Si O Si OH OH CH 3 Si O Si O Si O CH 3 CH 3 CH 3 CH 3 CH 3 currentpoint
Cl
Si
CH 3 CH 3
La phase stationnaire sera lie au support par des interactions hydrophobes.
b) La phase stationnaire
Elle doit tapisser de faon aussi uniforme que possible les parois des pores du support. Cette phase doit tre trs peu miscible avec la phase mobile et sa viscosit doit tre la plus faible possible pour que les phnomnes de transfert de masse et de diffusion soient facilits. L'imprgnation peut tre ralise de plusieurs faons : par vaporation du solvant : la phase stationnaire est dissoute dans un solvant appropri puis mlange avec le support. Le solvant est ensuite limin par vaporation. par filtration du solvant : dans ce cas le solvant de la phase stationnaire est limin par filtration.
r Jean-Louis CUQ -
page 45
par percolation : la phase stationnaire est "dissoute" dans un solvant volatil, puis on la fait percoler travers la colonne ; on chasse le liquide interstitiel puis on vapore le solvant par un courant gazeux. En faisant varier la concentration des solutions de phases stationnaires, on peut faire ainsi varier l'paisseur du film sur le support.
VI.2.2. La phase mobile
Le choix de la phase mobile est empirique ; on se laisse guider par la notion de polarit que l'on peut estimer par le paramtre de solubilit d'Hildebrand (tableau 7). Ce paramtre d rsulte de la combinaison de trois principales interactions entre le solut et le solvant Tableau 7. Classement des solvants selon le paramtre de solubilit d'Hildebrand (G = grand). SOLVANT
p
6 7 7,1 7,3 7,4 6,7 8,2 8,6 8,9 8,1 7,6 9,2 6,8 7,2
o
0 0 0 0 0 2 0 0 0 3 4 0 5 2,5
a
1 0 0 0 0 2 0 0,5 0,5 0,5 3 0,5 2,5 4
d
0 0 0 0 0 0 0 0 0 3 0 0 0 4
SOLVANT Pyridine Ethanol Actonitrile Acide actique Dimthylsulfox. Mthanol Ethanolamine Ethylne glycol Formamide Eau
10,4 11,2 11,8 12,4 12,8 12,9 13,5 14,7 17,9 21
p
9 6,8 6,5 7 8,4 6,2 8,5 8 8,3 6,3
o
4 4 8 7,5 5 G G G G
Perfluoroalcanes 6 isooctane 7 n-Pentane 7,1 n-Hexane 7,3 n-Heptane 7,4 Ether dithylique 7,4 Cyclohexane 8,2 Ttrachlorure carb. 8,6 Tolune 8,9 Chloroforme 9,1 Ttrahydrofurane 9,1 Benzne 9,2 Actone 9,4 Propanol 10,2
5 0 5 5 2,5 0 5 7,5 G G G G 0 7,5 G G G G
- interactions de dispersion (forces de London) mesures par p. Les solvants avec des p leves donneront des interactions fortes avec des drivs polarisables (composs armatiques, composs halogns ou soufrs de masse molaire leve). - interactions diples-diples mesures par . Les solvants avec des leves donneront des interactions fortes avec des soluts fort moment dipolaire (nitriles, sulfoxydes, amides, drivs nitrs). - interactions hydrogne rsultant soit d'un pouvoir accepteur de proton mesur par p soit d'un pouvoir donneur mesur par a . Les solvants de a lev donneront des liaisons hydrogne avec des soluts donneurs (phnols, acides carboxyliques etc...) et les solvants de p lev avec les soluts accepteurs (amines, sulfoxydes etc...). Chacun des 4 paramtres (a , p, o, d,) pris isolment ne peut rendre compte de la polarit de tel ou tel solvant envers un solut donn: c'est la combinaison () qui permet d'apprcier le mieux cette proprit. On commence en gnral par choisir un solvant de telle sorte que les facteurs de capacit des divers soluts soient compris entre 1,5 et 10. En CLL classique (phase stationnaire polaire) la force d'un solvant augmente avec sa polarit et il suffit de diminuer d pour que les facteurs de capacit diminuent. C'est l'inverse en CLL polarit de phase inverse. Les couples phases stationnaire-phase mobile les plus employs en chromatographie liquide-liquide sont indiqus dans le tableau 8.
r Jean-Louis CUQ -
page 46
Tableau 8. Quelques systmes utiliss en CLL phases stationnaires phases mobiles
Chromatographie liquide classique
'propionitrile carbowax trithylne glycol thylne glycol dimthylsulfoxyde thylne diamine eau polyamide
pentane, hexane, isooctane idem + 20 % de chloroforme, THF actonitrile ther di n-butylique isooctane hexane n-butanol hexane/mthanol
Chromatographie liquide polarit de phases inverse
cyanothylsilicone dimthylpolysiloxane heptane polymre hydrocarbon squalane mthanol/eau actonitrile/eau mthanol/eau mthanol/eau actonitrile/eau
VI.3. LA CHROMATOGRAPHIE D'ECHANGE D'IONS
La phase stationnaire est un support insoluble contenant des groupements chargs. Ceux d'un certain signe sont fixes car lis chimiquement, les autres de signe oppos sont mobiles. Ces derniers peuvent tre changs rversiblement avec d'autres ions de mme charge sans aucun changement de la partie insoluble. Les groupes ioniques fixes dterminent le type et la force de l'changeur. Le nombre et l'accessibilit dterminent la capacit. Pour des composs monovalents (ion du solut et contre-ion) le processus d'change peut tre simplement dcrit par les quilibres suivants :
change d'anions
avec
B+ Y- + X+ + -
KXY currentpoint currentpoint- + YB+ X
-
K XY =
B X
B Y X
on tire [ B+X-] que lon porte dans DX
change de cations
A- M+ + NH+
KNM currentpoint currentpoint + + M+ A- NH
KXY et KNM sont les constantes d'quilibre de la raction d'change. X- et NH+ sont les soluts Y- et M+ sont les contre-ions. B+ et A- sont les groupements ioniss fixs sur la phase stationnaire. Ka currentpoint Pour des acides on a XH currentpoint H+ + X-
r Jean-Louis CUQ -
page 47
avec
Ka =
et pour les bases
on tire [XH] que lon porte dans DX currentpoint currentpoint N + H+ NH+
H X XH
Le coefficient de distribution D (rapport des concentrations du solut charg dans la phase stationnaire sur la somme des concentrations du solut charg et non charg dans la phase mobile) sera gal : change d'anions change de cations
DX =
B X
-
+ -
X + XH
= K XY .
B Y Y
-
+ -
. 1+
1 H Ka
+
DN =
NH A
+ +
N + NH
= K NM .
MA M
+
+ -
. 1+
1 Ka H
+
D est proportionnel la constante d'qulibre de l'change (KXY, KNM) et au nombre de groupes ioniss de la matrice ([B+Y-], [A-M+]. D est inversement proportionnel la concentration en contre-ion ([Y-], [M+]) dans la phase mobile. D est fonction du pH et du pKa. Le mcanisme d'lution est schmatis sur la figure 20.
-Echangeur d'anions et contre-ions interchangeables
+ -+ + + + +
Echangeurs de cations et contre-ions interchangeables
Figure 20 . Chromatographie d'change d'ions. Types d'changeurs et mcanisme de l'lution. 1. l'changeur d'ions est en quilibre aves les contre-ions (O). Les ions changer et sparer pntrent dans l'changeur 2. les contre-ions ont t changs avec les ions sparer 3. on applique un gradient d'lution 4. la dsorption d'1 des constituants intervient , il est lu 5. les ions restants sont changs et lus 6. l'changeur est rgnr.
r Jean-Louis CUQ -
page 48
VI.3.1. Principaux types de groupements fonctionnels
Les groupements les plus utiliss sont de type sulfonate (- SO3-), phosphonate (- PO2-3) et carboxylate (- COO-) pour les changeurs de cations et de type ammonium quaternaire (- N+ R3), tertiaire (- N+HR2) ou secondaire (- N+H2R). Les groupements de type sulfonate ou ammonium quaternaire sont des changeurs d'ions forts, les autres des changeurs faibles. Ainsi la fraction charge d'changeurs de type sulfonate ou ammonium quaternaire n'est pas influence dans une zone large de pH (figure 21) tandis que les changeurs faibles prsentent des variations importantes de leurs ionisations entre pH 4 et 8.
fraction molaire charge
A
0 0
B
7
D
14
A changeur de cations fort B changeur de cations faible
pH Figure 21. Variation de la charge de groupements fonctionnels utiliss en chromatographie d'change ionique en fonction du pH
C changeur d'anions faible D changeur d'anions fort
Certains groupements possdent des proprits complexantes vis--vis de certains cations mtalliques (phosphonate, aminodiactate).
VI.3.2. Principaux types de matrice
Il existe des matrices de type organique (polystyrne, mtacrylate, dextrane) cellulosique ou silice. Les rsines de type polystyrne (BARNES N., Internat. Lab., oct 1993, 16, P4-P8) rsultent de la polymrisation de vinylbenzne avec comme agent de pontage du divinylbenzne (4 8 %). Ces matrices sont synthtises dans une phase apolaire solvante des monomres disperse sous forme dmulsion dont la taille des particules disperses est parfaitement contrle.
A
CH CH2 n n CH CH2
CH
CH2
CH
CH2
CH CH2
CH CH2 styrne ou vinylbenzne CH CH2
CH
CH2
CH
CH2
CH CH2 CH CH2 divinylbenzne
Structure dune matrice de phase stationnaire de type polystyrne. A = groupement fonctionnel ionisable (souvent SO3H).
r Jean-Louis CUQ -
page 49
Les matrices dont le support est la silice ont par exemple les structures suivantes :
Si SI Si OH CH 3 O Si CH2 CH2 CH 3 OH SO3 H Si SI Si OH CH3
(CH2 )3 O CH2 CH CH2 NH 2 OH
Il existe des changeurs dions bras mobiles (J.Chromatogr., 1988, 443, 73-83) qui permettent une sparation non dnaturante de macromolcules (protines, acides nucliques, particules virales etc) et qui confrent la phase stationnaire une proprit exclusive dchangeur dions, les interactions hydrophobes, hydrogne, - tant pratiquement ngligeables. Les interactions entre une protine globulaire et cette phase ou une phase classique sont schmatises sur la figure 22.
phase stationnaire classique protine globulaire pH > pHi
phase stationnaire bras mobiles
figure 22 . Effets dune changeur danions classique et tentaculaire sur la conformation dune protine globulaire . Ces phases stationaires sont obtenues en utilisant des supports sphriques de silice ou polymriques comportant de trs larges pores et de diamtre voisin de 5 m, particules la surface inactive. Les changeurs danions de type dimthylaminothyle, faiblement basiques, ou les changeurs de cations de type carboxyle ou sulfonate ont t lis au support par lintermdiaire de polymres linaires greffs. La configuration flexible des groupes dchangeurs contribue alors diminuer le trajet et donc la diffusion du solut mais aussi datteindre plus de groupements acides sur la molcule protique ou dacide nuclique ce qui contribue augmenter la slectivit. Un exemple de chromatogramme de protines sur changeurs de cations et danions bras mobiles ou classique est schmatis sur la figure 24 .
r Jean-Louis CUQ -
page 50
B C
1
absorbance 280 nm absorbance 280 nm
30 B
60
30 D
60
3
absorbance 280 nm
C E absorbance 280 nm
4
G
30 temps de rtention (minutes)
60
30 temps de rtention (minutes)
60
figure 24 . Sparations de protines par change de cations. A = chymotrypsinogne A, B = cytochrome C, C = lysozyme.
1 : changeur bras mobiles sur une phase de type gel porteuse de groupements carboxyles (colonne de 15 cm x 0,5 cm). 2 : changeur sulfon tentaculaire dp = 5 m, colonne de 5 cm x 0,5 cm, lution par gradient (dihydrognophosphate de sodium 20 mM pH 6 avec Na Cl de 0 1 M en 60 min). 3 : changeur traditionnel de type TSK-CM. Pour 1 et 2 lution par gradient (tampon actate pH 5; 10 mM avec Na Cl de 0 1 M en 100 min, dbit 1ml.min-1. 4 : sparation de conalbumine (D), ovalbumine (E), -lactoglobuline A ( F) et -lactoglobuline B (G) sur changeur danions TMAE, colonne de 15 cm x 1 cm, lution par gradient (piprazine 20 mM pH 6 et Na Cl de 0 O,3 M entre 10 et 45 min), dbit 1,2 ml.min-1. VI.3.3. Influence du pH de la phase mobile
Il s'agit du paramtre principal de rtention . Ainsi pour un acide faible, une augmentation de pH se traduit currentpoint AH currentpoint A- + H+ par un dplacement de l'quilibre vers la gauche, l'acide AH tant peu (ou pas) retenu. Avec les acides amins, on utilise une colonne changeuse de cations (polystyrne sulfon). On se place initialement dans des conditions de pH ( 2) pour lesquelles tous les acides amins sont sous la forme + NH3 - CH - COOH R Si on injecte sur la colonne des solutions tampon de pH croissant, les acides amins seront lus en fonction de leur pHi. En effet, quand le pH de la phase mobile est gal au pHi l'acide amin se trouve sous forme de switterion non charg, donc non retenu. + NH3 -CH - COOR Avec les composs polyhydroxyls (oses, diholosides etc...) on ralise un complexe avec l'acide borique, complexe charg ngativement.
r Jean-Louis CUQ -
page 51
H H3BO3 + 2 H OH H C O C OH H C O B O C H O C H + + H + 3 H 2O
Ces complexes peuvent tre spars sur rsines changeuses d'anions (tampon borate avec gradient de pH de 7 10).
VI.3.4. Influence du type et de la concentration en contre-ion
La constante d'quilibre de l'change reflte l'affinit du solut ionique ou du contre-ion pour l'changeur. En gnral les changeurs d'ions ont une grande affinit pour : - les ions possdant une forte charge - les ions avec un petit volume d'hydratation. Il est possible de classer les ions en fonctions de leur affinit pour l'changeur, donc de leur "force d'lution". Ainsi avec un changeur d'anions du type ammonium quaternaire on a : citrate > ClO4- > SO24 > oxalate > I- > NO3- > Br- > SCN- > Cl- > formiate > actate >OH- > FAvec les rsines changeuses de cations de type sulfonate on a : Ce3+ > Ba2+ > Pb2+ > Sr2+ > Ca2+ > Ni2+ > Ca2+ > Cu2+ > Co2+ > Zn2+ > Mn2+ > Ti+ > Ag+ > Cs+ > Rb+ > K+ > NH4+ > Na+ > H+ > Li+. Quand le solut et le contre ion sont compltement chargs, la pente de la droite log D(N ou X) en fonction de la concentration en contre-ion est en gnral gale a/b, avec a valence du solut, b valence du contre ion. Quand la concentration en contre-ion augmente (X-) B+ Y- + X-
currentpoint currentpoint B+ X- + Y
on dplace l'quilibre vers la droite.
VI.3.5. Effet de la temprature et de solvants organiques
Si on augmente la temprature de la colonne, la rtention du solut diminue. De plus la viscosit de la phase mobile diminue ce qui permet d'augmenter le dbit. L'addition de solvants organiques la phase aqueuse mobile affecte la rtention. En effet, les effets secondaires de la matrice (adsorption hydrophobe dans le cas de polystyrne et permation) jouent un rle prpondrant dans la distribution des ions et plus particulirement des ions organiques.
r Jean-Louis CUQ -
page 52
VI.3.6. Analyse par chromatographie dchange dion avec suppresseur de phase (SAARI-NORDHAUS R., ANDERSON J.M., Intern. Lab., 1994, 18, P4)
Cette mthode est utilise pour supprimer la conductance apporte par lluant sous forme de contre-ion. Les phases suppresseurs sont des matrices de type polystyrne et sont souvent prsentes sous forme de fibres creuses. Ces phases permettent le dosage danions avec une rsolution remarquable. NaOH
siemens
F- Cl-
SO4 --
NaF ; NaCl , Na 2SO 4 dans Na OH analyse en sortie
temps got H+ 0,5
S HF , HCl , H2 SO4 dans H 2O
F-
Cl-
SO4 --
fibre creuse NaX
temps
NaOH Na+ H2 OHH2 O
anode
O2
cathode
H+
H+ + X-
H2 O
HX
dtecteur
Cette mthode est utilisable pour analyser des anions ou des cations .
VI.4. LA CHROMATOGRAPHIE D'EXCLUSION VI.4.1. Principe
La chromatographie d'exclusion, ou gel filtration ou gel permation est fonde sur la rtention des molcules de solut en fonction de leur taille en raison de leur pntration dans les pores, remplis de solvant, d'une phase stationnaire approprie. Si on suppose que les molcules de solut ne prsentent aucune affinit pour les parois de la phase stationnaire particulaire, les grosses molcules ne pourront pas pntrer dans les pores ; elles migreront plus rapidement que les petites molcules qui peuvent, quant elles, pntrer dans un plus grand nombre de pores (figure 25
r Jean-Louis CUQ -
page 53
molcules de solut
Figure 25. Chromatographie d'exclusion. Schma de "trajet " d'un solut de masse molaire leve (S) et d'un solut de masse molaire faible (s). Les grosses molcules sont lus par un volume Vo de solvant qui correspond au volume entre les "grains" de la phases stationaire. Les petites molcules pntrent dans le rseau du gel et sont lues par un volume Vt gal au volume total de solvant contenu dans la colonne. La sparation est donc fonde ici, non sur des interactions physicochimiques avec la phase stationnaire, mais sur la dimension des molcules en solution. Le volume de rtention VR des molcules X est gal :
VR = Vo + K . F. Vi
Vo = volume total de la phase mobile ou volume mort , Vi = Vt - Vo - Vmatrice du gel Vi = volume total de la phase stationnaire F est la fraction du volume poreux de la phase stationnaire accessible aux molcules X. F est compris entre 0 et 1. K coefficient de distribution des molcules X K = Cs / C M, Cs et CM tant les concentrations respectives de X dans la phase stationnaire et dans la phase mobile. Les reprsentations de Vo, Vt et Vt - Vo sont schmatises sur la figure 26.
Vo
Vt-Vo
Vt
Figure 26. Diagramme reprsentatif de Vt et Vo En chromatographie d'exclusion pure K est gal 1 et on a : Il y a 2 cas limites importants :
1) F = O. Les molcules sont totalement exclues de la phase stationnaire et on a VR = Vo. Toutes ces molcules mergent ensemble de la colonne aprs le passage d'un volume de phase mobile gal au volume interstitiel de la colonne. VR = Vo + F. Vi
r Jean-Louis CUQ -
page 54
2) F = 1. Tous les pores de la phases stationnaire sont accessibles aux molcules considres et on a VR = Vo + Vi.
Entre les deux limites, VR varie linairement en fonction de la fraction du volume poreux accessible. Ainsi, chaque valeur de VR correspond thoriquement une taille de molcule elle-mme porportionnelle la masse molaire (figure 27).
log (masse molaire)
F = 0 (exclusion totale) permation slective F = 1 (permeation totale)
VR Vo+Vi Vo Figure 27 : Relation entre la taille ou la masse molaire et le volume de rtention. 0
Si VR > Vt cela implique K > 1 : il se superpose alors un autre mcanisme de rtention (adsorption change d'ions). Quand K = 1, les isothermes sont des droites et les pics sont des courbes de Gauss parfaites. Tous les constituants du mlange chromatographi sont lus dans un volume infrieur Vt. VR = Vo + F V VR - V0 F= Vi avec V = V - V - V En pratique on remplace Vi par Vt - Vo ; on obtient alors :
K av = VR - V0 Vt - V0
i t o matrice du gel
Kav est la fraction du volume du gel stationnaire o diffuse une molcule donne. Pour des composs de densit et de forme molculaire identiques, il existe une relation sigmodale entre les valeurs de leur Kav et le logarithme de leur masse molaire. Ces courbes prsentent dans une partie importante, une relation linaire entre Kav et log MM (figure 28); cette relation est du type :
Kav = A log (.M) + B
A et B sont des constantes, viscosit intrinsque (ml.g-1)
Kav 1 A
0,5
Log (poids molculaire)
Figure 28. Variation du Kav en fonction du logarithme de la masse molaire A gel "grande" taille" de pores, faible rticulation B gel "moyenne" taille de pores, rticulation plus leve.
r Jean-Louis CUQ -
page 55
Un exemple de courbe de calibration pour la chromatographie d'exclusion est donn sur la figure 29.
log MM 3
palmitate de vitamine A rythromycine rserpine actate de cholestrol prednisone probecenid propylparaben mthylparaben aspirine acide salicylique
2,5
2 0 5 10 VR (ml)
Figure 29. Courbe d'talonnage pour la chromatographie d'exclusion Colonne Styragel 100 A, 30 cm x 0,7 cm ; solvant THF 2 ml / min-1.
VI.4.2. Diffrents types de gel existent actuellement
On distingue les gels mous, semi-rigides et rigides. Seul ce dernier peut tre utilis avec des vitesses de phase mobile leves (FPLC par ex.). Certains gels sont rendus apolaires par modification des fonctions hydroxyliques. L'alcoylation des polymres de dextrane conduit des gels stables en prsence de solvants organiques ce qui permet le fractionnement des substances insolubles dans l'eau (LH). Ces gels sont vendus sous forme de grains secs et doivent tre mis gonfler avant utilisation dans une colonne dans la phase mobile approprie en doivent tre dgazs avant utilisation pour chasser les bulles dair qui pourraient rester prisonnires de la matrice.
a) Gels mous :
Ce sont des polymres faible taux de pontage pour lesquels le gonflement est important. Il s'agit le plus souvent de dextrane rendu insoluble dans l'eau par raction l'pichlorhydrine (figure 30).
currentpoint
O CH2 H OH OH O H H H OH H OH agent de pontage OH H O H O CH2 OH H O H CH2 H C OH CH2 OH H OH O H OH CH2 O O CH 2 OH H H O H O CH2 H C CH2 O H H H OH O CH 2 OH H O H H OH O OH O CH 2 H OH CH2 OH H O H H OH O CH CH 2 Cl pichlorhydrine O
currentpoint
Figure 30. Structure partielle dun gel de dextrane.
r Jean-Louis CUQ -
page 56
La rsistance mcanique augmente avec le taux de rticulation tandis que le diamtre des pores diminue. Il existe des gels dont le polymre est du polyacrylamide et des gels mixtes composs d'acrylamide et de dextranes. L'agent de pontage est le N-N'-mthylne bisacrylamide (figure 31).
currentpoint
CH2 NH O NH C O C CH CH2 CH CH2 O CH2 H CH2 CH C NH CH2 NH C O CH CH2 CH CH2 OH OH O H H H OH O H CH2 O O O H H H OH O H OH CH2 O H H H OH O CH2 OH H CH2 O OH O OH O H H OH H OH CH2 CH2 H O CH2 OH O H O CH H OH H H O CH2 OH H O H OH H O
currentpoint
Figure 31. Gel mixte dextrane-acrylamide
b) Gels semi-rigides
- Gels de polystyrne. Ce type de gel possde des proprits remarquables et ne gonfle pratiquement pas. Il peut tre utilis dans la plupart des solvants sauf : l'eau, les alcools, l'actone, l'acide formique. Il est utilisable en haute pression. - Gel d'actate de polyvinyle. - Gels mixtes (dextrane-acrylamide) taux de rticulation lev. Plus le taux de rticulation augmente et plus la rsistance aux contraintes mcaniques augmentent : ces gels supportent des P relativement leves.
c) Gels rigides
Ils sont constitus par des silices poreuses ou des verres poreux. Les colonnes HPLC du type TSK correspondent des phases stationnaires de ce type (poreuses, mais rsistantes aux dformations mcaniques et donc utilisables sous des dbits et pression levs). Un chromatogramme de protines solubles de haricot est indiqu sur la figure 32. Ce type de chromatographie (associ ou non des phnomnes dchanges dions) est qualifi de FPLC (Fast Protein Liquid Chromatography).
absorbance 206 nm
0,5
10
20
t R (minutes)
figure 32 . FPL Chromatogramme par exclusion des protines de pois
r Jean-Louis CUQ -
page 57
VI.4.3 Principales applications de ce type de chromatographie :
a) La sparation de groupes de molcules de masses molaires trs diffrentes
Le gel est choisi pour que les plus grosses molcules aient un Kav voisin de 1 - dessalage : les protines et les polypeptides peuvent tre dessals ou spars de substances de faibles masses molaires avant concentration. - extraction du phnol dans la prparation d'acides nucliques. - interruption de ractions entre macromolcules et ractifs de faible masse molaire. - extraction de produits, cofacteurs ou inhibiteurs d'enzymes
b) Dtermination de la masse molaire c) Dtermination des constantes d'quilibre
Pour le dessalage ou la sparation de protines / petites molcules, la comparaison de diverses mthodes est indique dans le tableau 9. Tableau 9. Comparaison de mthodes de sparation protines / petites molcules
Critre Simplicit Efficacit du dessalage Scurit Filtration sur gel Facile Approche habituellement 100 % Absolument sr Dialyse conventionnelle en sacs de cellulose Facile Peut atteindre 100 % si le procd est conduit son terme Sacs de dialyse susceptibles de se fissurer ou de casser Faible Trs longue, le procd peut prendre plusieurs jours Adsorption gnante pour de faibles concentrations Habituellement impossible Faible Mthode des fibres creuses Facile Environ 95 %
Les fibres peuvent fuir ou se casser Cher 95 % de dessalage en 20 minutes au mieux Peut atteindre 70 % au moins pour de faibles concentrations Habituellement impossible Faible
Cot Dure
Faible 100 % de dessalage obtenu en 15 minutes Habituellement proche de 100 %
Rendement des protines
Rcupration des petites molcules Dilution
Habituellement proche de 100 % Dpend du volume et de la concentration de l'chantillon Habituellement 1.000 utilisations au minimum. Les colonnes peuvent tre frquemment tre utilises 1 2 ans sans qu'il soit ncessaire de les refaire.
Dure d'utilisation
Une seule utilisation
Jusqu' 50 utilisations mois.
ou 4
r Jean-Louis CUQ -
page 58
VI.5. LA CHROMATOGRAPHIE CHIRALE
(ARMSTRONG D.W., Current issues in HPLC Technology, LC.GC International, april 1998, 22-31). Cest W. PIRKLE (PIRKLE W.H. and HYUN M.H., J. Chromatography, 1985, 322, 309) qui est un des principaux initiateurs des ces mthodes analytiques. Traditionnellement cest la sparation des nantiomres qui est considre comme le problme le plus difficile rsoudre en chromatographie. Avant 1980 aucune solution ntait disponible. Entre 1980 et 1990 de nombreuses mthodes ont t mises au point pour atteindre de tels objectifs. Ce sont surtout les sparations des acides amins D et L qui ont suscit le plus de travaux. DAVANKOV et al. (Advances in Chromatpgraphy, Giddings et al. Eds, M. Dekker, New York, 1983, vol 22, p 71) ont ainsi propos une phase stationnaire avec de la L-proline greffe. En prsence de Cu2+ dans la phase mobile, les interactions qui stablissent varient en fonction de lnantiomre.Il en est de mme avec des phases stationnaires silice greffe C8 en prsence de compos amphipolaire C asymtrique. O N OCu2+ OC O H NH 2 R
H
Si O CH3
Si
(CH )7 2 (CH )17 2 CH3
CH3 H
Si O
CH3
(CH )7 2
NH
COO_
++ 1/2 Cu
Un exemple de sparation de D et L tyrosine et de D et L tryptophane est prsent ci-dessous.
TYR = 1,34
TRP =1,11
20
40 temps d'lution (min)
La phase mobile est constitue dun mlange eau/mthanol (85/15) contenant 2 mM de CuSO4. Le dbit est de 1 ml/ min. La colonne de 15 x 0,4 cm est remplie de particules de 5 m de diamtre. La dtection est ralise 254 nm.
En dehors de cette mthode de sparation par change de ligand, des phases stationnaires chirales ont t dveloppes comme par exemple les phases polythers (formule indique ci-dessous) ou encore les phases qui contiennent des molcules complexes comme certains antibiotiques glycopeptidiques (ticoplanine par exemple). Elles permettent entre autre la sparation des D et L acides amins.
O O O O H O H N+ O H O R acide amin ther chiral en couronne C H O-
r Jean-Louis CUQ -
page 59
LIPKOWITZ K.B. (Intern. Lab., 1993, 15, 8-12) a propos une modlisation molculaire en chromatographie chirale qui aboutit : k = A + i bi. k avec A = constante, bi moindre carr des coefficients de rgression multiple. Dans cette quation tous les changes phase stationnaire - analyte sont pris en considration. Selon les conformation relatives des deux structures, la somme de ces changes sera variable en fonction des possibilits striques dtablissement de liaisons et k sera diffrent selon la structure spatiale des analytes. Avec des phases comportant des carbones asymtriques, il sera alors possible de sparer des analytes qui sont des diastroisomres. currentpoint
B H O C O O R C H C N H R
phase chirale
N C H
Les changes sont qualifis par PIRKLE de : donneur dlectron : analyse de sulfoxydes, lactames etc accepteur dlectron : analyse damines, alcools, acides amins thiols etc. Le schma danalyse des composs isomres est le suivant : isomres constitution stroisomres (conformation) gomtriques nantiomres (diastroismres)
currentpoint
complexe avec un phase stationnaire phase stationnaire driv chiral achirale chirale Il existe une classification des phases chirales base sur les mcanismes impliqus dans la sparation des nantiomres qui est la suivante :
type 1 2 3 4 5
description Pirkle polymres hlicodaux cavit change de ligand protines
chimie slecteurs chiraux varis liaisons ioniques celluloses et drivs cyclodextrines polymtacrylates polyacrylamides SAH , SAB glycoprotine
mcanisme liaisons H interactions - diple - diple liaisons H complexes d'inclusion complexes d'inclusion
interactions multiples hydrophobes
r Jean-Louis CUQ -
page 60
VI.5. LA CHROMATOGRAPHIE D'AFFINITE
C'est en 1910 qua t observe l'adsorption slective de l'amylase sur un amidon insoluble. Cette technique, encore qualifie de bio-chromatographie ou encore de bio-reconnaissance, permet de purifier presque toutes les molcules biologiques selon leur fonction ou leur structure chimique individuelle. Il s'agit d'une chromatographie d'adsorption dans laquelle la molcule purifier est adsorbe spcifiquement de faon rversible sur un ligand immobilis sur la phase stationnaire. Le facteur de purification est trs lev car lnergie change est trs leve et rsulte de ltablissement de nombreuses liaisons souvent de nature diffrente et coopratives. De nombreuses sparations ont pu tre accomplies en une seule tape. Elle est utilise pour : - purifier des produits prsents dans ses mlanges biologiques - purifier la forme active d'un produit de son driv inactif - sparer des traces de molcules biologiques de grandes quantits d'impurets.
VI.5.1. Principe
Un ligant biospcifique est fix par liaison covalente une matrice (dextrane, agarose) sans perdre son affinit pour le produit purifier.
A
A Li P KD
Li
P
K= Li-P Li P
A Li
agent de pontage ligand produit purifier constante de dissociation
Tout compos peut servir de ligand pour purifier les produits qui sont mme de se fixer sur lui :
enzyme anticorps lectine acide nuclique
analogue de substrat, inhibiteur, cofacteur : : : antigne, virus, bactrie, cellule polyoside, glycoprotine, rcepteur de surface cellulaire squence complmentaire de bases, histone, acide nuclique, polymrase rcepteur, protine porteuse protine de surface.
hormone, vitamine : cellule
r Jean-Louis CUQ -
page 61
VI.5.2. Couplage et ligand
Il existe plusieurs mthodes de couplage. La plus utilise fait appel la carbodiimide
O COOH NH2
+ R1 N C N R2 carbodiimide
NHR1 O C NR2
Li
O C NH
Li
matrice
Li = ligand
Il est possible de fixer sur la matrice des thiols (type glutathion -2-pyridyl disulfure)
S N
+ SH R
fixation
R' SH S S R
R'
lution
+ R SH
RSH peut tre une protine, un peptide etc. Par cette mthode chromatographique, on peut par exemple purifier les immunoglobulines en utilisant la protine A comme ligand. Quelques enzymes et protines possdent une affinit pour un colorant bleu (bleu Cibacron F3GA) et plus particulirement les kinases, les dshydrognases et les enzymes contenant des groupements adnyl (620 sur 2000 enzymes).
O NH2 SO2ONa R1 R2 NH O NH NH SO 2ONa N N N O
R1 = H ou SO2 ONa R2 = SO2 ONa ou H
matrice
Dans ce type de chromatographie, il y a reconnaissance et interactions souvent multiples (hydrogne, hydrophobe, ionique, transferts de charge - ) entre deux molcules : le solut et la phase stationnaire. Des essais de prparation de phase stationnaire par empreinte molculaire sont souvent couronns de succs. Pour ce faire, la molcule sparer est complexe avec des monomres ractifs et le complexe molcule-monomres est plac dans un milieu contenant un polymre que lon rticule alors. On dbarrasse ensuite le polymre phase stationnaire de la molcule empreinte. Les monomres sont souvent des composs vinyliques ou acryliques, des styrnes ou des silices tandis que lacrylate dimthylique de lthylne glycol constitue la phase rticulante. Cette mthode permet par exemple la sparation des racmiques dacides amins.
r Jean-Louis CUQ -
page 62
CH2 H COOH NH3+
+ COO
CH
CH2 CH2 H COOH NH3+
polymre
+ polymre
D-phnylalanine
COO-
CH
CH2
empreinte de D-PHE D-PHE
CH2 H COOH NH3+
extraction de la molcule de D-PHE
polymre
COOCH2 H NH3+ COO-
CH
CH2
L-PHE
VII. EQUIPEMENTS UTILISES EN CHROMATOGRAPHIE LIQUIDE
Le schma gnral dun systme chromatographique en phase liquide est indiqu sur la figure 33. Ce systme comprend : - lluant qui peut constituer une phase mobile de composition constante (systme isocratique) ou variable en fonction du temps (systme gradient ) - le systme de pompage pompe unique (systme isocratique ou gradient basse pression) ou multiple (amortissement ou sytme gradient haute pression) - un systme dinjection - une colonne (thermostate ou non) prcde ou non dune pr-colonne - ventuellement un racteur post-colonne - un ou plusieurs dtecteurs avec enregistreur et intgrateur
r Jean-Louis CUQ -
page 63
PHASE MOBILE
diffrence de pression hydrostatique faible
solvant : rservoir(s)
Dgazage par entrainement l'hlium ou par ultra-sons
filtre 0,45 m
systme de pompage
isochratique ou gradient basse ou haute pression drivatisation pr-colonne dispositif automatique passeur automatique phase stationnaire drivatisation postcolonne
injecteur pr-colonne colonne
thermorgulation
collecteur de fractions
dtecteur(s) enregistreur intgrateur traitement des donnes
figure 33. Schma dun systme de chromatographie en phase liquide. Un systme de chromatographie liquide doit satisfaire un certain nombre de contraintes dont deux sont primordiales : - l'lution travers des colonnes contenant des phases stationnaires de fine granulomtrie qui ncessite des pressions leves -lobtention dune bonne reproductibilit des temps de rtention qui ncessite un dbit de la phase mobile constant. VII.1. DISPOSITIFS D'OBTENTION DE GRADIENTS Les plus anciens sont des rservoirs communicants. Actuellement les plus rpandus sont obtenus par des systmes grs par microordinateurs et programmation. Llution peut tre obtenue au moyen de dispositifs permettant de raliser des gradients basse pression de polarit, de pH, de force ionique, de concentrations en divers composs (dispositifs chambres communiquantes, figure 34)
7 6 5 4 3 2 1 C/Co 1 1 2 4 3 5 6 C/Co 7
0,5
Ve / Vt
figure 34 . Schma dun dispositif dlution par gradient chambres communicantes
r Jean-Louis CUQ -
page 64
Dans ces dispositifs, les variations de concentration C (par rapport la concentration initiale Co) dun compos donn plac dans un des compartiments en fonction du volume lu (pomp) sont indiques sur la figure 34. A un volume dlution donn (ou un temps dlution donn) on peut, avec ces systmes, valuer lachambre dont lincidence est dominante et celles intervenant un degr moindre dans llution. Ainsi, au dbut de lanalyse cest la composition de la chambre n1 qui est dterminante tandis qu mi-analyse, ce sont dans lordre les chambres n4, puis n3 et 5 et enfin n3 et 6 qui par leur composition influenceront le plus llution. En choisissant judicieusement la composition de ces chambres pour obtenir des variations de pH, force ionique, concentration en mthanol et en thiodiglycol permettant la sparation (figure 35), il est possible de raliser en quelques heures ou moins (avec des colonnes de particules de rsines changeuses de cations de type polystyrne sulfon de 5 15 mm de diamtre et 5 50 cm de longueur, le plus souvent 50C et avec des dbits de tampons citrates voisins de 1 ml.min-1 ) la sparation complte de tous les acides amins .
mthanol (%) pH 6 mthanol 10 + Na pH 1,2 0,5 + Na (M) thiodiglycol (%)
5 2,5 0 0 0,2 0,5 Ve /Vo 1 0
SER GLU PRO GLY ALA CYS VAL
LEU NorLEU TYR PHE
MET LEU
ASP
CYSSO3H
figure 32. Variations des paramtres de sparation des acides amins par chromatographie dchange ionique (gradient). Le chromatogramme obtenu est schmatis ci-aprs.
cysSO3 H met O2 met O asp thr ser gly glu ala met val cys leu ile norleu tyr phe lys his arg
THR
TRP
HIS
pro
trp NH4
temps d'lution
- Llution peut galement tre obtenue par des systmes trois luants (ou plus) dont les mlanges, grs en fonction du temps par ordinateur, conduisent des gradients dlutions qui permettent
ARG
LYS
r Jean-Louis CUQ -
page 65
des variations des paramtres de llution tels que le pH, la force ionique, la polarit (valeur de o, nature des luants etc.) (figure 36 ). Si le mlange est ralis avant le systme de pompage, le systme est qualifi de systme gradient basse pression . Une seule pompe suffit alors llution, mais il est ncessaire de bien mlanger les diffrents luants primaires dans des systmes dont le volume mort peut constituer un handicap. Si le gradient dlution est ralis par autant de pompes quil y a dluants primaires (au maximum 3 actuellement), le systme est qualifi de gradient haute pression. Les dbits des diffrentes pompes sont rgls lectroniquement de telle sorte que la somme de leurs dbits respectifs soit gale au dbit dlution choisi. A B C A B tampons d'lution
lectrovannes mlangeur
pompe
SYSTEME BASSE PRESSION
SYSTEME HAUTE PRESSION
injecteur boucle exyterne colonne dtecteur
enregistreur
figure 36 . Schma des systmes basse et haute pression danalyse en HPLC VII.1. LES POMPES Il existe des pompes pression de gaz, mais les plus utilises sont les pompes lectriques de type seringue (jusqu 20-50 bars) et surtout les pompes de type piston (figure 34). Cest avec ce dernier type de pompe que des dbits constants trs faibles (0,1 mL.min-1 ) ou trs grands (plusieurs mL.min-1) sont accessibles des pressions qui peuvent atteindre environ 500 bars. Les pistons de ces pompes sont en saphir, en tantale ou en matriau rsistant et inaltrable, les pistons en acier inoxydable tant relativement sensibles la corrosion. Il faut rappeler ici que les solutions salines sont particulirement corrosives et quil est bon de rincer lensemble du systme pour viter ces phnomnes. Par ailleurs si des sels sont prsents dans la ou les phases mobiles, lvaporation de ces dernires conduit leur cristallisation ce qui est particulirement gnant quand ce phnomne se produit
r Jean-Louis CUQ -
page 66
dans des zones sensibles (cellules de dtection, vannes dinjection, etc) difficilement accessibles au nettoyage.
sortie piston vannes billes (saphir)
partie motorise
joint torique entre
Pompe de type seringue ( 50 500 ml dplacs)
ressort de rappel
sortie piston vannes billes
came excentrique
joints toriques Pompe piston unique
entre variation de la pression en sortie
sortie
pompe A
pompe B
entre de la phase mobile
variations de P pour la pompe A
variations de P pour la pompe B
variation globale de P
Pompe 2 pistons opposs fonctionnant en opposition de phases
figure 37 . Schma de quelques pompes utilises en chromatographie en phase liquide Il est important aujourdhui de choisir des pompes pouvant assurer des dbits de lordre de quelques L. min-1 afin de pouvoir les utiliser avec les nouvelles colonnes (capillaires de moins de 0,5 mm de diamtre et contenant des particules de phase stationnaire de 1 m de diamtre).
VII.3. LES INJECTEURS Il faut absolument viter de dissoudre les soluts dans un solvant plus luant que la phase mobile. Il est souhaitable de dissoudre les soluts dans le liquide vecteur. Il existe deux types dinjection : - par seringue - par vanne (boucle dchantillonnage). Ce type dinjection automatisable ne permet pas datteindre lefficacit maximale. Il existe des vannes boucles internes et des vannes boucles externes (cF V.1.3.4 figure 6).
r Jean-Louis CUQ -
page 67
Dans lanalyse des acides amins, il faut que la phase solvante de lhydrolysat contenant les acides amins analyser ait un pH et une force ionique quivalents ceux du tampons dlution au temps 0 pour une sparation par chromatographie dchange ionique mais aussi possde une polarit gale celle de la phase mobile au temps 0 de lanalyse dans le cas dune sparation par chromatographie en phase inverse. VII.4. LA COLONNE (cf chapitre V.1.3.2.) Les frits limitant la colonne ont des porosits comprises entre 0,5 et 5 m. Linconvnient majeur des frits de faible porosit est le colmatage qui se traduit par des augmentations importantes de pression. Lutilisation de particules de 5 m requiert la prsence de frits de 0,5 m. VII.5. LES PARTICULES SUPPORT DE PHASE STATIONNAIRE Comme indiqu dans le tableau ci-aprs, la silice reste le matriau de base le plus utilis en HPLC. Ses caractristiques sont largement dveloppes dans le chapitre VI.1.2.1. matriau gel de silice polymre organique polymtacrylates polystyrne-DVB polythylne glycol alumine autres graphite, carbone zirconium hydroxyapatite utilisation estime (%) 75 20
1 4
Le plus utilis des polymres organiques reste le polystyrne rticul avec du divinyl benzne. De porosits variables, ils sont en gnral stables dans une gamme de pH comprise entre 1 et 14. Leurs caractristiques principales sont indiques dans le chapitre VI.3.2. Parmi les polymres commercialiss comme phase stationnaire on peut citer : les polystyrnes divinylbenzne, le polydivinylbenzne, les polymtacrylates, le polybutadine, le polyvinylpyrrolidone, le polydextrane, la cellulose, le polyhydroxymtacrylate, lthylvinylbenzne, alcools polyvinyliques alkyls, polyosides, polyhydroxythyl aspartamide, chlorure de polyvinyle, triactate de cellulose, mthylne bis acrylamide, polythylne glycol, agarose rticul, etc.. Les matrices organiques sont moins utilises que la silice comme support de phase inverse. Nanmoins elles permettent des sparations chromatographiqes dans des conditions de pH pour lesquelles la silice nest pas utilisable. La figure 22 schmatise des rsines de type polystyrne ou silice qui peuvent tre :
r Jean-Louis CUQ -
page 68
microporeuses
( trs rticules par le DVB ) ; ces rsines ne gonflent pas , leur slectivit est leve mais les transferts de masse y sont mauvais (cintique lente) (peu rticules) ; ces rsines ont des canalicules et une surface interne importante. Une grosse molcule peut pntrer l'inrieur (cintique moyenne)
micropores
macroporeuses
macropores 1 m pelliculaires L'paisseur de rsine sur la bille de verre est de l'ordre de1 m (cintique d'change trs rapide)
film changeur d'ions microsphres
40 m
bille de verre : particule de silice superficiellement poreuses La rsine changeuse d'ions est fixe sur des microsphres de silice , elles-mmes fixes sur des billes de verre . L'paisseur de la couche de rsine est infrieure 1 m
figure 22. Diffrents type de particules de phase stationnaire en chromatographie liquide. Les rsines pelliculaires ont une capacit d'change de 260 1000 fois plus faible que celle des rsines poreuses. Par contre leurs HEPT sont environ 10 fois plus faibles. La taille des pores des particules varie entre 4 et 300 nm. La plupart des colonnes proposes sont replies de particules pour lesquelles la porosit est denviron 8-12 nm. Les particules macroporeuses (perfusives) permettent des chromatographie prparatives ncessitant des dbits levs avec peu de perte de charge. Leur efficacit est en gnral infrieure celle des particules normales . Actuellement la tendance est lutilisation de particules dont le diamtre est compris entre 3,5 et 5 m. Ces supports permettent un bon compromis entre la facilit dusage et loptimisation de lanalyse (temps, rsolution etc.). Il sagit le plus souvent de partcicules aux dimensions polydisperses. La distribution gaussienne des diamtres prsente des carts-types relativement faibles comme schmatis sur la figure ciaprs :
nombre de particules
4 5 6 diamtre en m Distribution de diamtre de particules de silice avec une valeur moyenne de 5 m
r Jean-Louis CUQ -
page 69
VII.6. LA NATURE DE LA PHASE STATIONNAIRE
(cf chapitre VI.1)
Le gel de silice a t substitu, partir des annes 1975, par des gels greffs et depuis 1984 ce sont les phases inverses qui dominent les applications de lHPLC. Plus de 95 % des analystes utilisent en HPLC, au moins de temps en temps, des phases inverses. Lutilisation relative en 1998 des diffrentes phases disponibles en HPLC est indique dans le tableau ciaprs :
mthode chromatographique
Utilisateurs relatifs (%)
mthode Utilisateurs chromatographique relatifs (%)
exclusion 6,7 phase aqueuse non aqueuse chirale interaction hydrophobe affinit partage autres 2,8 1,1 0,6 0,2 0,3 3,5 3,2
phase inverse 50,4 C18 24 C8 15,9 phnyl 7,1 C4 2,3 C1-C2 1,1 phase normale 24,1 cyano 8,9 silice 8,5 amino 4,7 diol 2,0 change d'ions 14 anion 7,4 cation 6,6
Linfluence de la mthode de prparation des particules de silice influence de faon trs significative les performances de la sparation Le grains de silice greffs avec des C8 C18 possdent encore des silanols libres qui sont neutraliss par raction avec du trimthylmonochlorosilane. La longueur des chanes greffes aliphatiques (phase inverse) influence la stabilit : plus le nombre de CH2 du greffon est lev et plus la stabilit aux pH infrieurs 2 et suprieurs 8 augmente ( C30 > C22 > C18 > C12 > C8 > C6 > C4 > C2 > C1). Le dopage de la silice par de laluminium qui ragit avec les silanols gminaux conduit une structure pseudo-zolithique stable jusqu pH 10.
VII.7. LES CONNEXIONS
Les connexions injecteur-colonne-dtecteur constituent un volume mort qui en diluant les soluts dans la phase mobile entranent une perte d'efficacit non ngligeable. Pour ne pas perdre plus d'une fraction
2 2
1+
de l'efficacit il faut que :
r .l
2 24 . . D M. VR
.N.D
r Jean-Louis CUQ -
page 70
r et l : rayon intrieur et longueur du tube DM : coefficient de diffusion du solut dans la phase mobile de dbit de l'luant. Ainsi pour r = 0,2 mm l doit tre infrieur ou gal 320 mm et pour r = 0,5 mm l Rappelons que la dispersion dans la colonne est gale :
=
2
8 mm.
tr
et que la largeur la base des pics est gale 4 .
TAYLOR a montr que la dispersion lie aux tubes de liaisons entre linjecteur, la pr-colonne, la colonne et le dtecteur tait gale : 4 2 dt . l . D = . DM
A est une constante, dt le diamtre intrieur des tubes de liaison, l leur longueur, D le dbit de la phase mobile et DM le coefficient de diffusion du solut dans la phase mobile. La prsence de cavits ou de volumes morts divers contribue aussi la dispersion.
VII.8. LES DETECTEURS (tableau 10)
Il existe actuellement 3 types principaux de dtection : - mise en vidence d'une proprit du solut directement ou aprs raction pr ou post-colonne avec un compos rvlateur (spectrophotomtre UV/visible, fluorimtres), - mise en vidence d'une modification de l'luant (conductimtre, rfractomtre), - sparation du solut de l'luant (couplage chromatographie - spectromtrie).
VII.5.1. Critres de choix
Les paramtres les plus importants considrer sont : 1.le bruit donn en units de la mesure du dtecteur. 2.la sensibilit : concentration pour laquelle le rapport signal/bruit est gal 2. 3.la drive exprime en units de la mesure par unit de temps. 4.le domaine de linarit : domaine de concentration de rponse linaire du dtecteur. 5.le volume de la cellule qui peut tre considre comme une chambre de mlange. 6.la possibilit d'utiliser un gradient dlution. Depuis 1971 lvolution de lemploi des divers dans les laboratoires a beaucoup volu comme le montre le tableau 10a ci-aprs :
r Jean-Louis CUQ -
page 71
Tableau 10a. Evolution de lutilisation (en %) des dtecteurs en CLS
anne
UV-visible
fixe var. Baret.
type de dtecteur
fluor. 12 10 12 7 1 Indice Electro Conduc- Spectro rfract. chim. -timt. masse 4 5 9 13 34 7 8 6 3 4 3 2 1,5 2 8 1,5 1 0,3 Diffusion 4 2 -
1991 1986 1981 1976 1971
63 69 66 73 61
10 21 25 48 46
43 10 43 5 41 24 15 -
Les principales caractristiques des dtecteurs sont indiques dans le tableau 10b. Tableau 10b. Principaux dtecteurs utiliss en chromatographie liquide.
type Dtecteur UVvisible UV-visible barettes diodes Fluorimtre Universalit +++ +++ + Limite de dtection 0,3 ng/ml 1 ng/ml 5 pg/ ml drivatisation Rfractomtre Dtecteur lectrochimique Dtecteur par conductivit Spectromtre de masse Diffusion de lumire +++++ ++++ slective d'ions ++++++ ++++++ 0,7 mg/ml pg/ml 0,1 ng/ml ng /ml 1 ng glucose + +++++ +++ + ++++++ Destructeur +/Identification de composs + ++ ++ facilit d utilisation +++++ +++++ Remarques et cot Le plus utilis peu coteux bande passante assez coteux
++++ ncessite souvent une ++ + ++ +/+ peu coteux cellule lecture dtecteur davenir limit aux ions trs cher dtecteur davenir
VII.5.2. Dtecteurs par absorption UV/visible
C'est le dtecteur le plus utilis. Le faisceau de longueur donde donne traverse une cuve dans laquelle circule l'effluent. Suivant la loi de BEER l'absorbance est dfinie par : Io A = log = . l . c I Io intensit incidente I intensit mergeante coefficient d'extinction molaire du solut
r Jean-Louis CUQ -
page 72
c concentration l longueur du trajet optique Le bruit est de l'ordre de 10-5 units d'absorbance, le domaine de linarit voisin de 104 et le temps de rponse est de l'ordre de 0,5 seconde. Il existe des dtecteurs barettes de diodes (35 100 diodes par barettes, 1 barette rfrence et 1 barette dosage), diodes sensibles des longueurs d'onde dfinies, en gnral de 2 nm en 2 nm (parfois 10); ces dtecteurs permettent de raliser des spectres d'absorption en des temps trs courts donc en cours de chromatographie (BERTOLIN M., Intern. Labor., oct 1991,44-50). Les rsultats sont prsentes sur des chromatogrammes en trois dimensions.
VII.5.3. Les fluorimtres l Certains soluts qui absorbent des photons h . C / ex , peuvent rmettre des photons dnergie h . C / m La longueur d'onde d'excitation ex est infrieure celle d'mission. L'intensit de fluorescence F mise par une solution de concentration C irradie par une radiation d'intensit Io est donne par : F = Q K Io (1 - 10-cl)
K est le rendement de collecte de la fluorescence Q fraction de radiation rmise. La dtection trs slective et trs sensible ncessite parfois une drivatisation pralable.
VII.5.4 Dtecteur diffusion de lumire
(WYATT P.J., Intern. Labor., 1993 ; STOCKWELL P.B. et KING B.W., 1991) Le solvant est nbulis par un courant de gaz inerte (azote) et vaporis une temprature infrieure ou gale 180C. Le solut dont la tension de valeur est suprieure celle de lluant reste ltat de brouillard et est amen par le gaz vecteur jusqu la chambre de dtection. La quantit de lumire diffuse mesure par un photomultiplicateur plac 120 par rapport la lumire incidente est proportionnelle la masse lue. Il sagit dun excellent moyen de dtection quand la masse molculaire est suprieure 200 Da.
VII.5.5. Dtecteurs lectrochimiques
Un potentiostat impose l'lectrode de travail un potentiel fixe par rapport celui de l'lectrode de rfrence tel que le solut dtecter soit oxyd ou rduit. Au cours du passage du solut dans la cellule, un courant d'lectrode proportionnel sa concentration circule entre l'lectrode de travail et sa contre lectrode.
VII.5.6. Dtecteurs rfractomtriques
On mesure en continu la diffrence d'indice de rfraction entre le solvant pur et l'effluent de la colonne. Ces dtecteurs surtout utiliss pour les glucides sont de loin les moins sensibles. Il faut veiller ce que leur cellule de mesure soit rince leau ou avec un solvant organique comme le mthanol aprs chaque srie de mesures car si la phase mobile contient des sels, ceux-ci risquent de cristalliser dans le systme et leut solubilisation est extrmement difficile.
r Jean-Louis CUQ -
page 73
VII.5.7. Couplage chromatographie liquide / spectromtrie de masse
Le couplage CL- SM permet dobtenir un systme analytique trs performant. Par chromatographie liquide il est possible de sparer nimporte quelles molcules en choisissant judicieusement le systme et les composs spars peuvent tre injects dans un spectromtre de masse qui donne des informations immdiatres sur la masse molaire et la structure .
a) Principe Le compos analyser est dabord vaporis basse pression. Ce transfert en phase gazeuse empche les molcules de lchantillon de ragir avec les autres et facilite lionisation qui est ltape analytique suivante. Le spectromtre dtermine la masse molculaire des composs en mesurant leurs rapports masse / charge ( m / z ). Lionisation de la molcule gnre des formes charges du compos qui sont diriges vers une srie de champs magntiques ou lectrostatiques. Plusieurs mthodes ont t dveloppes pour ioniser le compos spar par chromatographie liquide . b) Mthodes dionisation Lionisation des molcules peut tre obtenue par un faisceau dlectrons dirig dans la phase gazeuse. Lnergie cintique de ces lectrons permet de rompre des liaisons covalentes intramolculaires. Les fragments obtenus donnent des informations permettant de structurer la molcule. Un des dsavantages de cette ionisation est que de nombreuses molcules ont des liaisons covalentes de faible nergie qui sont rompues sans donner dions dtectables correspondants, ce qui empche alors de dtermijner la masse molculaire. Lionisation chimique est employe pour minimiser la fragmentation tout en produisant lion dtectable . Le produit est ionis par transfert de proton partir de gaz (mthane) ionis par des lectrons acclrs , ce gaz tant inject automatiquement linterface . c) Linterface chromatographie liquide / spectromtrie de masse Elle permet de transformer le liquide correspondant la phase mobile en un gaz dans un milieu pression trs faible. De nombreuses interfaces ont t imagines (ceinture mobile, thermovaporisateur, injecteur direct de liquide, transport par un faisceau de particules, bombardement atomique rapide et continu etc.). Dans un thermovaporisateur la phase mobile contenant le solut analyser est chauffe dans le liquide vaporisateur et ensuite vaporise dans la zone sous vide; il se forme un fin brouillard. Si la phase mobile contient des ions NH4+, actate) , quelques microgouttes peuvent porter des charges. Sous vide solvant svapore rapidement et les molcules de solut sont dsolvates et associes la charge. Le rsultat est une molcule charge isole de la phase mobile. Pour des soluts non ioniss le systme de vaporisation est associ un filament source dlectrons qui dcharge des ions pour faciliter la formation dions ou encore une lectrode de fragmentation qui gnre des fragments en augmentant la vitesse des collisions intermolculaires. d) Diffrents types danalyseurs de masse Ainsi ionises, les molcules sont diriges vers des zones champs lectrique et magntique. Il existe ce niveau quatre types danalyseurs de masse:
- secteur magntique. Dans ce systme, pour un champ magntique donn et une vitesse donne de la particule, seuls les ions molculaires dune masse et dune charge donnes peuvent passer dans le dtecteu . Pour valuer la gamme de masses molculaires prsentes, lintensit du champ est modifie plusieurs fois afin de dtecter tous les ions prsent . Ce dtecteur a une rsolution de 0,001 U de masse molculaire. - lanalyseur quadrupole est bas sur linteraction de la molcule ionise avec un champ lectrique oscillant. En faisant varier le potentiel appliqu aux lectrodes, seuls les ions dune masse donne oscillent dans une direction stable et atteignent le dtecteur. Les autres molcules entrent en collision avec les lectrodes et ne sont pas dtectes. Lanalyse des masses prsentes est obtenue en faisant varier le potentiel
r Jean-Louis CUQ -
page 74
appliqu aux lectrodes et en dterminant pour un potentiel donn les ions prsents. Avec ce systme le balayage des masses est rapide (1 seconde) et ce systme est utilisable en couplage CPV ou CPL. - lion trap analyseur utilise aussi des interactions dans un champ lectrique. Alors que dans lanalyseur quadrupole lion avec une trajectoire stable est dirig vers le dtecteur, dans ce systme, ce sont les ions stabiliss dans la cellule qui sont analyss. - lanalyseur de temps de transfert mesure le temps ncessaire un ion pour atteindre le dtecteur (50 100 sec). Un exemple de couplage chromatographie liquide / spectromtrie de masse est donn sur la figure 39.
absorbance 280 nm
B A
colonne C18 , 10 cm L , 0,4 Di luant tampon actate pH 4 0,01 M / mthanol ( 80 / 20 ; V/V ) dbit 0,8 ml / min
temps de rtention (minutes)
100 50
160
204
currentpoint
m/z 32 76 261
NH2
0 abondance relative (%)
CH2 CH COOH N H
50 0
47 91 276
NH2 CH2 CH CO NH
CH2 COOH CH3
N H
50
NH2 CH CH2 CH CO NH COOH
C currentpoint
0
m/z
N H
figure 39 . Couplage chromatographie liquide - spectromtrie de masse
VII.5.8. Les racteurs post-colonne
Une raction chimique peut tre ralise entre un chromophore et les soluts dtecter. Plusieurs types de racteurs sont utiliss : capillaire, segmentation de flux Pour lanalyse des acides amins le systme suivant est souvent utilis.
r Jean-Louis CUQ -
page 75
O OH OH O O O
+R
NH2 CH COOH
ninhydrine
O
COOH NH CH R O + H2O O
OH
COOH N CH R + H2O
H NH2
O H N CHR N CH2 R O +CO
HO
2
+ R-CHO
O
H O
+ ninhydrine
O OH H O hydrindantine O
O N
+NH 3
+ ninhydrine
pourpre de Ruheman compos violet NH 3
O
OH OH
O O
O O
OH NH2
Les acides amins lus individuellement sont soumis en continu une raction colore permettant leur dosage. Il sagit le plus souvent dans la chromatographie dchange ionique de la raction la ninhydrine qui est ralise pH 5,5 en prsence de mthylcellosolve, sous azote et chaud. Parmi les nombreux drivs qui se forment certains sont violets, dautres jaunes ou encore rouges, leur formation dpendant entre autre de la nature de lacide amin. La mesure de labsorbance plusieurs longueurs donde (570 et 440 nm ) aide l identification des acides amins lus. Le schma du racteur est indiqu sur la figure 40. Pour viter que des phnomnes de diffusion nlargissent les pics, le flux est segment par de lazote.
bain d'huile 2 4 95C
bobines de mlange
luat ninhydrine azote sulfate d' hydrazine
1 3 5
rfrigrant
pompe pristaltique
dtecteurs colorimtriques 440 nm 570 nm dgazeur
enregistreur intgrateur
figure 40 . Dosage continu des acides amins par raction la ninhydrine La pompe pristaltique est une pompe galets anime dun mouvement de rotation trs prcis. Les tubes de pompe ont des diamtres internes parfaitement calibrs donc des dbits parfaitement contrls .
r Jean-Louis CUQ -
page 76
galets azote
V
tube de pompe
La ralisation de ractions chimiques en continu avec ce type de racteur repose sur le principe suivant : si la raction en batch ncessite 3 ractifs A , B , C en quantits x , y et z, la raction en tube se fera avec des tubes de pompes permettant des dbits de x , y et z ml.min-1 , avec x/x = y/y = z/z. Si la raction ncessite un chauffage, une bobine est immerge dans un bain dhuile, sa longueur permettant de contrler le temps de passage. Aprs raction, le flux segment est dgaz par diffrence de dbits et un vent vertical, le dgazage tant ncessaire la mesure de labsorbance. Les cuves des spectrophotomtres de mesure en continu ont des trajets optiques compris entre 1 cm et 0,2 cm, leur volume tant compris entre 500 l et 5 l .
VIII. QUANTIFICATION
Aprs avoir obtenu des pics bien rsolus, il faut relier, pour chaque compos, le signal obtenu sur le dtecteur sa concentration dans l'chantillon analys. Sur le papier enregistreur on observe un pic gaussien (ou plus ou moins gaussien). L'aire du pic est proportionnelle la quantit injecte. A= A R
+ -
R . dt
aire du pic rponse du dtecteur
mi = Ki Ai
mi Ai Ki masse de solut dtect aire de son pic coefficient de rponse.
VIII.1. LA MESURE DE L'AIRE d'un pic peut s'effectuer :
1) par intgration lectronique 2) par pese du pic dcoup 3) par triangulation (figure 3, p 6) A = h x x 1,066 avec h hauteur du pic
hauteur mi-hauteur
4) par mesure de la hauteur des pics 2 tR N = 5,54 (mi = Ki Ai mi = Ki.hi.i 1,066)
r Jean-Louis CUQ -
page 77
m=K. h.
2,35 . t R
. 1,066
Cette mthode n'est utilisable qu'avec tR et N-2 constants (dbit parfaitement constant). Cette mthode est valable en chromatographie en phase gazeuse pour les pics troits.
VIII.2. MESURE DES COEFFICIENTS DE REPONSE Ki et de mi
Elle peut tre effectue - par talonnage direct - par mesure relative en rapportant la surface du pic du solut un compos de rfrence. Ki n'est pas le coefficient de rponse du dtecteur mais celui de l'appareillage tout entier, colonne comprise. La dtermination des concentrations peut se faire : a) par normalisation interne On mesure la proportion relative des aires en admettant que l'on connat Ki. La teneur d'un compos est Ai . K i m i (%) = . 100 iAi . K i b) par talonnage externe Cette technique n'est valable qu'avec une excellente qualit d'injection mref = K A ref On injecte une masse mref de compos talon en solution (Vrf inject). m rf m rf = Arf On injecte un mme volume de solution contenant le compos (me) et on mesure me. mi = K. Ai soit m rf mi = . Ai Arf c) par talonnage interne On introduit dans le mlange analyser un compos (talon interne) qui n'interfre pas avec les constituants du mlange et qui possde un k' voisin du ou de soluts doser. Avec cet talon interne on connat m ei K ei = Aei Si on a introduit m'ei dans le mlange analyser on doit trouver l'analyse une aire gale A'ei = Kei/mei. Si on trouve une aire diffrente (A"ei) c'est qu'il y a eu une perte (le plus souvent) de produit (par exemple au cours de la prparation de la solution). Cette "perte" est alors compense et on a : m rf A" ei mi = . Ai . Arf A' ei La rptabilit est la mesure de l'intervalle dans lequel sont trouvs des rsultats obtenus par un mme oprateur sur un mme appareil en oprant de faon identique.
r Jean-Louis CUQ -
page 78
La reproductibilit est la mesure de l'intervalle dans lequel sont trouvs des rsultats obtenus par des oprateurs diffrents sur des appareils diffrents mais avec un mode opratoire identique. La prcision caractrise par l'cart-type reprsente les carts par rapport la moyenne connue ou inconnue. La justesse est la valeur des carts entre la moyenne des mesures et la valeur relle.
VIII.3. INFLUENCE DES CONDITIONS OPERATOIRES SUR L'ANALYSE QUANTITATIVE
L'aire du pic A est gale A=
+
R . dt
(R rponse du dtecteur)
A = K C.dt. La rponse du dtecteur est fonction de la concentration C du solut. Si Q est le dbit de la phase mobile m = CdV = CQ dt Si le dbit de la phase mobile est constant on a : m = Q . C dt or A A C . dt = m=Q K K et Toute variation du dbit entranera une variation de l'aire d'o la ncessit de possder de trs bonnes pompes.
IX. TRANSPOSITION CHROMATOGRAPHIE LIQUIDE EN COLONNE SUR COUCHE MINCE
La chromatographie d'adsorption en colonne prsente certains avantages par rapport la chromatographie d'adsorption sur couche mince : - dure de sparation plus courte - analyse quantitative prcise - utilisable des fins prparatives - automatisation aise. La CCM est beaucoup plus facile mettre en oeuvre et peu coteuse. De plus, avec un mlange inconnu, il est possible de pratiquer une sparation sur CCM avec rvlation universelle (carbonisation par chauffage aprs pulvrisaton d'acide sulfurique). Les donnes bibliographiques sont trs nombreuses. En CCM on caractrise la migration d'un solut par son RF
RF =
distance parcourue par le solut distance parcourue par le front du solvant
Le facteur de capacit k' est WS k' = K VM WS masse de phase stationnaire volume de phase mobile VM K coefficient de distribution On a par ailleurs v v' = 1 + k' v' vitesse linaire du solut
r Jean-Louis CUQ -
page 79
v vitesse linaire du front du solvant Les distances parcourues sont proportionnelles leurs vitesses linaires d'o : v' 1 1 RF = = = v 1 + k' WS 1+K VM Si les phases mobiles et stationnaires en colonne et sur couche sont identiques K sera le mme dans les deux cas. Soit Ktr le coefficient de transposition WS colonne VM K tr = WS ' 1 CCM k cc = k tr( - 1) VM RF Pour des substances donnes il est alors possible, en connaissant leur RF sur couche mince de dterminer leur temps de rtention sur colonne : TR = to (1 + k'cc)
X. COMPARAISON AVEC LA CHROMATOGRAPHIE EN PHASE GAZEUSE
20 % environ des substances organiques connues sont justiciables de la CPG sans modification pralable de l'chantillon. La CPG prsence des limitations dans trois cas : - substances peu volatiles (mM > 300) - substances sensibles une lvation mme modre de la temprature (la plupart des substances d'intrt biologique) - substances ionises. La CPL est souvent plus efficace que la CPG pour 3 raisons : - en CPG il n'existe que des interactions solut-phase stationnaire alors qu'en CPL il y a des interactions additionnelles avec la phase mobile. - les phases stationnaires sont plus varies en CPL (change d'ions, exclusion). - la temprature est moins leve en CPL qu'en CPG. Or les interactions molculaires diminuent quand la temprature augmente. La CPL manque par contre de dtecteurs aussi universels que les dtecteurs catharomtre ou ionisation de flamme. D'autre part l'appareillage est plus complexe donc plus onreux. La CPG est une mthode simple, souvent plus rapide et plus sensible que la CPL. De ce fait, les deux mthodes ne sont pas concurrentes mais complmentaires.
r Jean-Louis CUQ -
page 80
XI - EXERCICES - ANNALES DEXAMENS
Sujet n 1.
La sparation dacides gras est effectue sur une colonne de 15 cm de longueur et 4,6 cm de diamtre interne dont la phase stationnaire est constitue de particules sphriques doctyl- silice de 4 m de diamtre. La phase mobile a la composition suivante :
CH3 CN V/V = 50
H3 PO4 O 22 28
1%
La colonne est rgule 35 et le dbit est de 1 ml.min-1. Le schma dun chromatogramme talon est donn sur C la figure 1 .
2 3 4 6 5 7 8 9 10
12 minutes
2 : acide linolnique 3: acide palmitolique 4: acide trans-hxadcnoque 5: acide linolique 6: acide trans-linolique
figure 1 . Schma du chromatogramme talon obtenu par injection de 10 l dune solution 0,1 mM dacides gras dans la phase mobile
(C18:3,cis) (C16:1,cis) (C16:1,trans) (C18:2,cis) (C18:2,trans) 7: 8: 9: 10: acide palmitique acide olique acide ladique acide starique (C16:O) (C18:1,cis) (C18:1,trans) (C18:O)
4 - Comment diminueriez-vous la dure de lanalyse ? ( o = 0,45 et 0,65 respectivement pour le ttrahydrofurane et pour lactonitrile). Quelles sont les limites vos propositions ? 5 - Quelle est la surface molculaire apolaire des acides gras de la srie C18 sachant que : ln k = 0,04 Sapolaire - 2 avec Sapolaire en 10-20 m2 . Interprtez les rsultats obtenus . 6 - Dans quelles conditions peut-on crire : (t - t ) Rs = N 2 1 2 ( t 2 + t 1) 7 - Quelle est la perte de charge , la viscosit de phase mobile tant gale 1 cP On donne
1 - Quel sont le type et le principe de cette sparation ? Quel est le rle de lacide phosphorique dans la phase mobile ? Pourquoi la sparation est-elle ralise 35 ? Expliquez lordre dlution des diffrents acides C gras ? Quel est le systme de dtection le mieux adapt ? 2 - La porosit interstitielle est gale 0,5 . Quel est le facteur de capacit de ces composs ? 3 - A quoi correspond le pic n 1 ?
dp . K = 180
o
(1 - )
r Jean-Louis CUQ -
page 81
Corrig n 1
1 - Il sagit dune chromatographie solide-liquide en mode isochratique dont la phase stationnaire est greffe apolaire ( C8) . Lacide phosphorique permet datteindre un pH infrieur au pKa des acides gras sparer ; les groupements carboxyliques sont alors sous la forme COOH ce qui permet la sparation sur la phase greffe apolaire en fonction du caractre hydrophobe de ces acides gras . La sparation est effectue 35 C dune part pour diminuer la viscosit apparente de la phase mobile et donc diminuer la perte de charge et dautre part pour favoriser la solubilit de certains des acides gras tudis et agir sur leur k. Les acides gras saturs ( C:16 et C:18) sont spars en fonction du nombre de leur CH2 . Plus celui-ci est lev et plus leur hydrophobicit est leve et donc leur k aussi . Pour un mme nombre nombre de CH2 et une mme conformation ( cis par exemple) , le facteur de capacit est dautant plus grand que le degr dinsaturation est faible . Pour un mme nombre de CH2 et un mme degr dinsaturation , k est plus lev pour la forme trans qui donne une molcule avec une surface apolaire plus grande que lisomre cis . Le systme le plus simple est la mesure de labsorbance 210 - 215 nm . La mesure de le diffusion de la lumire est galement possible . 2 - Volume de la colonne Vt = L . section = 15 . 3,141. (0,46)2 / 4 = 2,49 cm3 VM = . Vt = 0,5 . 2,49 = 1,25 cm3 to = VM / D = 1,25 min k =( tR - to) / to
4 - Une diminution de la dure danalyse est possible en diminuant o (donc k) ; ceci est ralisable en augmentant les proportions dactonitrile et surtout de ttrahydrofurane dans la phase mobile : risque de perte de rsolution. La dure danalyse peut tre rduite en augmentant v : risque de voir P augmenter 5 - Calcul de la surface molculaire apolaire ( en 10-20 m2) des acides gras en C18 : acide starique lnk = 2,106 Sapol = 102,7 acide ladique lnk = 2,072 Sapol = 101,8 acide olique lnk = 1,998 Sapol = 100 acide trans-linolique lnk = 1,686 Sapol = 92,2 acide linolique lnk = 1,515 Sapol = 87,9 acide linolnique lnk = 1,05 Sapol = 76,3 Les acides cis , cis-cis ou cis-cis-cis ont une structure replie qui leur confre une surface apolaire dautant plus petite ( en donc un k bas) que le nombre de doubles liaisons est grand . Les isomres trans occupent un volume plus grand que les isomres cis (voir Strutures des acides gras p2).Lhydrophobicit dune double liaison est infrieure celle de la liaison . 6 - La rsolution est donne par : 2 ( t 2 - t 1) Rs = ( 2 + 1) On a :
3 - Le pic 1 correspond au pic dinjection
acide linolnique (4,82 - 1,25) / 1,25 = 2,86 acide trans-hxa (6,35 - 1,25) / 1,25 = 4,08 acide trans-linol. ( 8 - 1,25 ) / 1,25 = 5,4 acide olique ( 10,47 - 1,25 ) / 1,25 = 7,38 acide starique ( 11,53 - 1,25 ) / 1,25 = 8,22
acide palmitolique (6 - 1,25 ) / 1,25 = 3,8 acide linolique ( 6,94 - 1,25) / 1,25 = 4,55 acide palmitique (9,53 - 1,25) / 1,25 = 6,62 acide ladique ( 11,18 - 1,25 ) / 1,25 = 7,94
) 2 Si les pics sont gaussiens et si on suppose que le nombre de plateaux thoriques est voisin pour les divers composs analyss , on a alors : 4 = . tR N ce qui permet dobtenir la relation propose . 7 - La perte de charge est gale :
P = . L . v K
o
N = 16 (
tR
K =
avec
dp 180
o (1 - )
2
On a : D v= .s soit v = (1/60) / 0,5. . (0,23)2 . -1 soit 0,2 cm.sec-1. v = 12 cm.min
r Jean-Louis CUQ -
page 82
Ko = (16.10-8 /180) . 0,5 =
4,44. 10-10
P = 10-2 . 15 . 0,2 / (4,44 . 10-10) = 3.10-2 / (4,44 . 10-10) = 0,675.10-8 baryes = 67,5 bars Structures des acides gras analyss :
currentpoint
CH3
COOH
acide starique
CH3
COOH COOH
acide ladique
CH3
acide olique
CH3
COOH
acide trans-linolique
COOH acide cis-linolique CH3 COOH acide linolnique CH3
r Jean-Louis CUQ -
page 83
Sujet n 2.
La structure chimique dune phase stationnaire qualifie de phnyl-ure et utilise en chromatographie LS est la suivante :
currentpoint
O Si O CH2 CH3 CH 2 CH2 H N C O H N CH3 H
Le diamtre des particules qui constituent cette phase est de 3 m. La colonne qui les contient a une longueur et un diamtre respectivement gaux 15 cm et 0,4 cm. Cette colonne est utilise dans un systme chromatographique complet pour analyser des acides amins et dipeptides de synthse (MET, GLY et GLY-MET). Un chromatogramme type obtenu est schmatis ci-dessous :
4 rponse du dtecteur injection 1 2 3 5
10
temps de rtention (min)
1) Expliquez le rsultat obtenu . 2) Proposez , en les justifiant , une ventuelle prparation de lchantillon et une composition satisfaisante de phase mobile permettant dobtenir un tel chromatogramme. 3) Quel est le systme de dtection le mieux adapt ? 4) Sachant que la porosit interstitielle est gale 0,5 quel est le dbit de la phase mobile ? Le volume de phase mobile compris entre linjecteur et la colonne dune part et la colonne et le dtecteur dautre part est gal 5,75 . 10-2 cm3. 5) Quelle est la perte de charge avec une phase mobile de = 1,24 cP ? On donne :
1 - 6) Quel est le facteur de capacit des acides amins et dipeptides analyss ? 7) Quelle est la rsolution entre les composs 2 et 3 et entre les composs 4 et 5 ? Discuter.
K =
dp 180
Corrig n 2
1 ) La phase stationnaire est une phase chirale (par la prsence dun C asymtrique) greffe en phase inverse. La glycine ne possde pas de C asymtrique et est lue sous forme dun pic unique n La mthionine se prsente 1. sous forme de deux isomres L-MET et D-MET (ou S et R ; nantiomres) et le dIpeptide GLY-MET sous forme de deux isomres : GLY-L-MET et GLY-D-MET (pics n 4 et 5 ou 5 et 4) 2) La prsence de deux fonctions (COO- et NH3+) ionisables nuit la sparation dans un systme en phase inverse. 2-1. Il est possible de saffranchir de lventuel effet de ces groupements en formant par exemple des drivs hydrophobes par condensation au niveau du NH3 ( raction avec par exemple l OPA ou la fluorescamine ou le PITC etc.) Dans ce systle de drivatisation prcolonne , la phase mobile sera tamponne ou acidifie un pH infrieur au pKa des composs analyss soit aux environs de 2 par exemple . Le temps de rtention des composs
r Jean-Louis CUQ -
page 84
sera dans ces conditions dautant plus lev que leur hydrophobicit sera grande et que le
VM = VT . soit 0,9425 cm3. Le volume mort total du systme est gal : VM = 0,9425 + 0,0575 = 1 cm3. A partir du chromatogramme on mesure to 2 min . Comme VM = to . D , on a : D = 1 / 2 = 0,5 cm3. min-1 . 5) La loi de Darcy donne : .L . v P = o K Ko = 4,445.10-10 v= D / .s soit v = 0,5 / 0,5 . 0,125 v = 8 cm.min-1 P = 55,6 bars 6) Valeurs de k : k = tR - to / to tR pic n 1 = 5 min (GLY) soit k = 3/2 = 1,5 tR pic n ( L ou D-MET ) = 8 min soit k = 6/2 = 3 2 tR pic n ( D ou L-MET ) = 8 ,3 min soit k = 6,3/2 = 3,15 2 2 tR pic n ( L ou D-GLY-MET ) = 10 min soit k = 8/2 = 4 tR pic n ( Dou L-GLY-MET ) = 10,6 min soit k = 8,6/2 = 4,3 2 7) La rsolution est gale : t R2 - t R1 Rs = 2 .
sera lev . La proportion de mthanol ou de THF ou dactonitrile dans la phase mobile est 2-2. Il est possible denvisager le blocage des fonctions COOH par estrification par exemple . 2-3 . Il est aussi possible de proposer de tamponner la phase mobile pH = 5,90 , pH correspondants sensiblement aux pHi des acides amins et peptides analyss . Dans ce cas il nest pas ncessaire de procder une drivatisation pralable . La composition de la phase mobile est envisager comme dcrit en 2-1 . 3) Si une drivatisationpr-colonne par des drivs fluorescents comme lOPA a t ralise , la dtection fluorimtrique simpose . En absence de drivatisation pr-colonne , une mesure de labsorbance 210 - 220 nm est possible . La drivatisation post-colonne est aussi possible . 4) Le volume de la colonne est gal ( D2 / 4 ) . 15 = 1,885 cm3. Le volume mort de la colonne est gal :
o de la phase mobile corrle au o .
soit
v = 0,133 cm . sec-1
Entre 4 et 5 : Rs = 2 ( 0,6 ) / 0,8 = 1,5 : la rsolution est complte car > 1 Entre 3 et 2 : Rs = 2 ( 0,3 ) / 0,8 = 0,75 : la rsolution nest pas excellente comme le montre le schma
2 + 1
Sujet n 3
1 En CLS , quelles sont les phases stationnaire et mobile utilisables pour sparer la dimthylamine, la gramine et )lhordnine. Donner, en fonction du systme choisi , lordre dlution de ces trois composs . 2 Quel est le type de dtecteur le mieux adapt aux systmes de phases choisis ? )3 Si pour analyser ces composs on utilise , en systme isochratique , une phase mobile qui contient du )naphtalne sulfonate de sodium 0,03M , quelle phase stationnaire et quel dtecteur sont alors ncessaires ? Quel doit tre le pH de la phase mobile ? Avec ce systme VT = 2 mL , la porosit e = 0,5, le dbit de la phase mobile = 1 mL.min-1 . 4 Quel est le facteur de capacit des composs analyss pour lesquels les tR sont respectivement de 3 , 6 et 9 ) min ? Quelle est la slectivit de la colonne ?
Corrig n 3
1) Il sagit damines R - NH2 sparables par :
r Jean-Louis CUQ -
page 85
- change dions des pH < pKa : lution avec un gradient de pH , changeurs dions de type sulfonate : lution du compos de pKa le plus faible vers celui du pKa le plus lev : gramine , hordnine , trimthylamine - chromato hydrophobe sur C18 ou C 8 : lution en fonction de lhydrophobicit (mthylamine , hordnine , gramine ) avec une phase mobile do adapt pH voisin ou suprieur au pKa - chromato par appariement dions ( avec un ractif paire dions du type sulfonate par exemple naphtalne sulfonate ) : lution idem quavec C18 mais avec une phase mobile contenant le ractif paire dions pH < pKa 2) Sans paire dions le dtecteur le mieux adapt est le spectrophotomtre = 200 nm , deux des composs cycliques absorbent aux environs de 260 - 280 nm Avec la paire dions sulfonate naphtalne une dtection fluorimtrique est possible ( sensibilit accrue) Diffusion de la lumire Dtecteur lectrochimique 3) Voir rponse en 1 et 2 4) = VM / VT do VM = 1 ml VM = to . D do to = 1 min
k' =
k mthylamine = 3 - 1 / 1 = 2 k hordnine = 6 - 1 / 1 = 5 kgramine = 9 - 1 / 1 = 8 Slectivit entre mthylamine et hordnine : = k2 / k1 = 2,5 Slectivit entre gramine et hordnine : = k2 / k1 = 1,6 Slectivit entre mthylamine et gramine : = k2 / k1 = 4
tR - to to
Sujet n 4
Des colonnes chromatographiques rcentes sont proposes avec 0,1 cm de diamtre interne et 5 cm de longueur . Ces colonnes sont garnies de particules sphriques de 0,8 m de diamtre et de porosit interstitielle gale 0,45 . Ces particules sont constitues de propyl silice et la phase mobile contenant du 1-hexane sulfonate a une viscosit gale 0,5 centipoise . 1) Quels sont les classes ou catgories de composs susceptibles dtre analyss au moyen de ce systme ? 2) Quels avantages et inconvnients prsente lutilisation de telles colonnes ? 3) Quel dbit de phase mobile doit-on fixer pour que la perte de charge soit gale 300 bars ? 4) Avec ce P , quel sera le to ? 5) Quel est la facteur de capacit dun compos lu 5 min ? 6) Quelle est la nombre de plateaux thoriques ? On donne :
dp Ko = . 180
P = Lv ko
3 2
(1 - )
HEPT = B .d p avec = 1,6 et B = 1
1) Appariement dions . Phase mobile B- , donc formation de paires dions avec les composs chargs + : il sagit en sciences des aliments surtout damines qui pH < pKa sont charges 2) Avantages : temps danalyse trs court , HEPT trs petit , N trs grand , volume dchantillon faible , utilisation dun faible volume de phase mobile
Corrig n 4
r Jean-Louis CUQ -
page 86
Inconvnients : systme dbit trs faible (nncessit de pompes de technologie avance ) , connexions , volumes morts , dtection trs difficile pour une masse trs faible de solut 3) dp = 0,8.10-4 cm , L = 5 cm , = 0,5 . 10-2 cP
0,64.10 0,091 -11 ko = . = 1,065 10 180 0,303
v= P . k o . L = 3.10 . 1,065.10 0,5.10 .5
2 8 11
-8
v = 0,128 cm/ sec D = v . s avec s = 0,785 . 10-3 cm2 D = 10-4 ml/sec = 6 l / min 4) to = L / v = 5 / 0,128 = 39 sec 5) tR = to ( 1 + k) 300 = 39 ( 1 + k ) do k = 6,7 6) HEPT = 1 ( 0,8 . 10-4)1,6 = 9.10-8 cm do N = L / HEPT = 5 / 9.10-8 = 5.107
Sujet n 5
1) Des colonnes chromatographiques de 0,1 cm de diamtre interne et 10 cm de longueur sont actuellement proposes garnies de particules sphriques de 1 m de diamtre et de porosit interstitielle gale 0,5. Ces particules sont constitues de butyl silice et la phase mobile contenant du 1-hexane sulfonate a une viscosit gale 0,5 centipoise .Quels sont les classes ou catgories de composs susceptibles dtre analyss au moyen de ce systme ? 2) Justifier le rle du pH . 3) Quels avantages et inconvnients prsente lutilisation de telles colonnes ? 4) Quel dbit de phase mobile doit-on fixer pour que la perte de charge soit gale 200 bars ? 5) Avec ce P , quel sera le to ? 6) Quel est la facteur de capacit dun compos lu 7 min ? 7) Quelle est la nombre de plateaux thoriques ? On donne :
Ko =
dp 180
(1 - )
HEPT = B .d p avec = 1,6 et B = 1
Corrig n 5
1) Appariement dions . Phase mobile B- , donc formation de paires dions avec les composs chargs + : il sagit en sciences des aliments surtout damines qui pH < pKa sont charges + 2) Se placer dans des conditions dee pH pour lesquelles lappariement dion est possible , par ex un pH pour lequel les amines sont charges + donc pH < pKa mais un pH > au pKa du sulfonate ( voisin de 1,5) 3) Avantages : temps danalyse trs court , HEPT trs petit , N trs grand , volume dchantillon faible , utilisation dun faible volume de phase mobile Inconvnients : systme dbit trs faible (nncessit de pompes de technologie avance ) , connexions , volumes morts , dtection trs difficile pour une masse trs faible de solut 4) dp = 10-4 cm , L = 10 cm , = 0,5 . 10-2 cP
-8 0,125 ko = 10 . = 2,78. 10-11 180 0,25 P . Ko v= . L do :
r Jean-Louis CUQ -
page 87
8 2,78 . 10-11 v = 2.10 . 10 0,5. 10-2
v = 0,111 cm / sec D = v . s avec s = (0,785 . 10-3 ) . 0,5 cm2 D = 4,36.10-4 ml/sec = 26 l / min 5) to = L / v =10 / 0,111 = 90 sec 6) tR = to ( 1 + k) 420 = 90 ( 1 + k ) do k = 3,67 7) HEPT = 1 ( 10-4)1,6 = 4.10-7 cm do N = L / HEPT = 10 / 4.10-7 = 2,5.107
Sujet n 6
De trs nombreux travaux de recherche ont t raliss pour prdire le temps de rtention de soluts au cours de leur analyse par chromatographie liquide . Quels sont les principaux paramtres prendre en considration dans le cas dune chromatographie dont la phase stationnaire est constitue de: 1) silice , 2) n-octadcyl silice . Une des dfinitions de lhydrophobicit des soluts a t propose par De Waterbeemd :
log10 P =
( Si solubilit du solut dans le 1-octanol et leau) ; les valeurs de log P sont disponibles pour de trs nombreux composs . En chromatographie en phase inverse on a : log10 P = a log10 K + b quation 1 ( K coefficient de distribution) , a et b tant des constantes . Quelles relations peut-on crire entre log P et k dune part ( k facteur de capacit) et logP et tR dautre part ? Deux composs A et B ont des solubilits respectivement gales 4 et 3,5 moles.l-1 dans le 1-octanol et de 0,4 et 0,5 mole.l-1 dans leau . Dans lquation 1 les valeurs de a et b sont pour les deux composs respectivement de 15 et 2,5 . Avec une colonne de 4 mm de diamtre et de 10 cm de longueur dont la phase stationnaire une porosit interstitielle de 0,5 ,un dbit de phase mobile de 1 ml.min-1 , quels sont les temps de rtention de A et B et quelle est la slectivit de cette colonne . Dans un systme isocratique phase mobile binaire ( eau : solvant organique ) avec une phase stationnaire du type octyl-silice, on dmontre exprimentalement que : log k = - S + log keau , quation dans laquelle k est le facteur de capacit , la fraction volumique du solvant organique , S la variation de pente lie un changement de 1 % de la concentration du solvant organique considr dans la phase mobile.Comment exploiteriez-vous cette quation ?
S 1-octanol Seau
Le temps de rtention dun solut en CLS est essentiellement fonction de la colonne ( L, nature et proprits et nature de la phase stationnaire ), de la phase mobile ( vitesse linaire , nature , temprature ), de la nature du solut. tR = ( 1 + k ) L / v , quation de Snyder log k = - a . o + b Silice : chromato dadsorption de soluts polaires ( k augmente avec la polarit du solut), tR dautant plus court que o est lev . n-octadcyl silice : chromato en phase inverse : adsorption de composs apolaires (k augmente avec lhydrophobicit du solut) , tR dautant plus court que o est faible . log10 P = a log10 K + b quation 1 log10 K = log10 k - log10 ( Vs / VM ) k = tR - to / to Pour rpondre remplacer les symboles par leur dveloppement A partir de lquation de De Waterbeemd :
Corrig n 6
log10 P =
pour A log10 P = 4 / 0,4 = 10 pour B log10 P = 3,5 / 0,5 = 7
S 1-octanol Seau
r Jean-Louis CUQ -
page 88
pour A 10 = 15 . log10 K + 2,5 soit log10 K = 0,5 pour B 7 = 15 . log10 K + 2,5 soit log10 K = 0,3 VT de la colonne D2 /4 . L = 1,26 cm3 = VM / VT = 0,5 k = K .VM / Vs = K pour A log10 K = 0,5 = log k soit k = 3,16 pour B log10 K = 0,3 = log k soit k = 2 = k2 / k1 = 3,16 / 2 = 1,58 VM = to . D do to = 0,63 . 1 = 0,63 min tRA = to ( 1 + k ) = 0,63 ( 1 + 3,16 ) = 2,62 min tRB = to ( 1 + k ) = 0,63 ( 1 + 3 ) = 1,89 min
donc VM = 0,5 VT = VS
Avec un systme isocratique phase mobile binaire (eau : solvant organique) et avec une phase stationnaire du type octyl-silice, on peut choisir k en fonction du pourcentage de solvant organique dans la phase mobile. log k = - S + log keau , quation dans laquelle k est le facteur de capacit, la fraction volumique du solvant organique , S la variation de pente lie un changement de 1 % de la concentration du solvant organique considr dans la phase mobile.Comment exploiteriez-vous cette quation ?
Sujet n 7
1 - De trs nombreux travaux de recherche sont raliss pour prdire le temps de rtention de soluts au cours de leur analyse par chromatographie liquide. Quels sont les principaux paramtres prendre en considration dans le cas dune chromatographie dont la phase stationnaire est constitue de: 1) silice, 2) n-octadcyl silice, 3) polystyrne sulfon, 4) silice-srum albumine humaine. 2 - Une des dfinitions de lhydrophobicit des soluts propose par De Waterbeemd est :
log10 P =
( Si solubilit du solut dans le 1-octanol et leau); les valeurs de log P sont disponibles pour de trs nombreux composs. En chromatographie en phase inverse on a : log10 P = a log10 K + b quation 1 ( K coefficient de distribution), a et b tant des constantes. Quelles relations existe-t-il entre log P et k dune part ( k facteur de capacit) et logP et tR dautre part ? 3 - Deux composs A et B ont des solubilits respectivement gales 4 et 3,5 moles.l-1 dans le 1-octanol et de 0,4 et 0,5 mole.l-1 dans leau. Dans lquation 1 les valeurs de a et b sont pour les deux composs respectivement de 15 et 2,5 . Avec une colonne de 4 mm de diamtre et de 10 cm de longueur dont la phase stationnaire une porosit interstitielle de 0,5 ,un dbit de phase mobile de 1 ml.min-1 , quels sont les temps de rtention de A et B et quelle est la slectivit de cette colonne . 4 - La largeur la base du pic A est de 10 secondes. Quelle est la HEPT ?
S 1-octanol Seau
1) Le temps de rtention dun solut en CLS est essentiellement fonction de la colonne (L, nature et proprits et nature de la phase stationnaire ), de la phase mobile (vitesse linaire, nature, temprature), de la nature du solut. tR = ( 1 + k ) L / v , quation de Snyder log k = - a . o + b Silice : chromato dadsorption de soluts polaires ( k diminue avec la polarit du solut), tR dautant plus court que o est lev. Si t augmente k diminue. n-octadcyl silice : chromato en phase inverse : adsorption de composs apolaires ( k augmente avec lhydrophobicit du solut ) , tR dautant plus court que o est faible. Polystyrnesulfon : change de cations. Deux paramtres importants : pH et . Silice-SAB : chromato chirale. 2) log10 P = a log10 K + b quation 1 log10 K = log10 k - log10 ( Vs / VM ) k = tR - to / to Pour rpondre remplacer les symboles par leur dveloppement
Corrig n 7
r Jean-Louis CUQ -
page 89
log10 P = a log10
tR V -1 . M +b t0 VS S 1-octanol Seau
3) A partir de lquation de De Waterbeemd :
log10 P =
pour A log10 P = 4 / 0,4 = 10 et pour B log10 P = 3,5 / 0,5 = 7 pour A 10 = 15 . log10 K + 2,5 soit log10 K = 0,5 pour B 7 = 15 . log10 K + 2,5 soit log10 K = 0,3 VT de la colonne D2 /4 . L = 1,26 cm3 = VM / VT = 0,5 k = K .VS / VM = K pour A log10 K = 0,5 = log k soit k = 3,16 pour B log10 K = 0,3 = log k soit k = 2 = k2 / k1 = 3,16 / 2 = 1,58
donc VM = 0,5 VT = VS
VM = to . D do to = 0,63 . 1 = 0,63 min tRA = to ( 1 + k ) = 0,63 ( 1 + 3,16 ) = 2,62 min tRB = to ( 1 + k ) = 0,63 ( 1 + 3 ) = 1,89 min 4) N = 16 ( tR / )2 et HEPT = L / N
HEPT =
10 t 16 . R
10 16 . 157 10
= 25 m
Sujet n 8
Des composs drivs du benzne sont analyss au moyen dun chromatographe haute performance quip dune colonne C8 (porosit = 0,5 , dp = 5 m, 15 cm de longueur et 3,9 mm de diamtre interne); le dbit de la phase mobile compose deau et disopropanol est de 1 mL.min-1. Ces drivs sont le nitrobenzne (NB), le 1,3dinitrobenzne (DNB), le 1,3,5-trinitrobenzne (TNB), le 3-nitrotolune (NT), le 2,4-dinitrotolune (DNT)et le 2,4,6trinitrotolune (TNT). Le chromatogramme obtenu est schmatis sur la figure 1.
TNB DNB TNT
rponse du dtecteur
NB DNT
NT
10
temps (min)
20
figure 1 : chromatogramme de drivs de benzne (C8) Expliquez le mcanisme de rtention et interprtez lordre dlution de ces divers composs. Quel type de dtecteur est bien adapt ce systme ? Si on reprsente le ln k en fonction du pourcentage disopropanol de la phase mobile on obtient pour le NB la droite schmatise sur la figure 2. Comment interprtez-vous cette droite ? Quel est le pourcentage disopropanol de la phase mobile utilise ?
r Jean-Louis CUQ -
page 90
3 ln k' 2
0 10 15 % isopropanol 20
figure 2. Variation du ln k en fonction du pourcentage disopropanol de la phase mobile pour NB La loi ln k = ln keau - S dans laquelle k est le facteur de capacit dans la phase mobile considre, keau est le facteur de capacit avec leau, S un paramtre de force du solvant et la fraction de ce solvant dans la phase mobile binaire, vous permetelle de contrler les temps de rtention des composs analyss. Avec un pourcentage disopropanol de 18 % dans la phase mobile, les variations du ln k en fonction de 103 / T (T : temprature absolue) sont indiques sur la figure 3.
3 ln k' 2
0 3,28 3,36 10 3 / T 3,44
figure 3. Variations du ln k en fonction de 103 / T pour le NB Discutez ce rsultat. A quelle temprature est ralise la chromatographie ? Quelle est la perte de charge en sachant que la viscosit de la phase mobile est de 1 cpoise ?
Corrig n 8
Les drivs benzniques analyss ont la structure suivante :
(NO2 ) NO 2 (NO2 ) CH3 NO2
(NO2 )
(NO 2 )
NB et (DNB et TNB)
NT et (DNT et TNT)
Il sagit dune chromatographie en phase inverse. Plus le nombre de NO2 est lev, plus la polarit du compos augmente et plus son temps de rtention est faible. Par la prsence du groupe mthyl, les drivs du tolune ont une hydrophobicit plus leve que les drivs du benzne; leur temps de rtention, toutes conditions tant gales par ailleurs, sont donc plus levs. Le noyau benznique absorbe dans lUV 254 nm : dtection par spectrophotomtre UV. Si le pourcentage disopropanol de la phase mobile augmente, o et lapolarit diminuent et donc le temps de rtention diminue. tR est li au facteur de capacit par tR = t0 ( 1 + k) . Si k diminue, ln k diminue et tR galement. VM = t0 . D = . VT do
t0 =
. VT 0,5 . 15 . 0,39 2 . = = 0,9 min D 4.1
r Jean-Louis CUQ -
page 91
Pour NB on a :
k' =
10 - 0,9 = 10,1 0,9
Par analyse graphique ln 10,1 = 2,32 soit % isopropanol 17,5 % Cest la loi de Vant Hoff qui montre que la temprature affecte lenthalpie de liaison des analytes sur la phase stationnaire :
0 0 V ln k' = - H + S + ln avec = S RT R VM
Pur 18 % disopropanol on a ln k = 2,3 soit 103/ T = 3,38 La temprature est de 23 C. La perte de charge est :
P =
h = 0,01 poise , L = 15 cm, v = D / s et s = s . v = 0,28 cm.sec-1 K0 = 0,07. 10-8 P = 60 bars
. L. v K0
Sujet n 9
Lanalyse des oses prsents dans du jus dorange est ralise au moyen dun chromatographe haute performance quip dune colonne de 15 cm de longueur , 0,39 cm de diamtre dont la phase stationnaire a une porosit de 0,5 et un dp de 5 m. Le dbit de la phase mobile compose dun mlange actonitrile / eau est de 1 mL.min-1 . La viscosit de cette phase mobile est de 1 cpoise. Le chromatogramme obtenu aprs injection de 100 L de jus est schmatis sur la figure 5 A.
S1 = 150 S2 = 150 S3 = 300 1 2
S1 = 300 S2 = 300
1 2
10
15 temps (min)
10 temps (min)
Figure 5 . Chromatogrammes de jus dorange (A) et de jus dorange trait linvertase (B). S est la surface relative des pics. Les coefficients de rponse des composs 1 , 2 et 3 sont identiques. Aprs action de linvertase sur le jus, le chromatogramme obtenu dans les mmes conditions analytiques est reprsent sur la figure 5B. Interprtez le rsultat. Quel est le mcanisme de rtention et quelle pourrait tre la nature de la phase stationnaire ? Interprtez lordre dlution des composs 1 , 2 et 3. Quel est le facteur de capacit du compos 3 ? Les variations du Ln k du compos 3 en fonction du pourcentage dactonitrile sont schmatises sur la figure 6. Comment interprtez-vous cette droite ? Quel est le pourcentage dactonitrile de la phase mobile utilise ?
r Jean-Louis CUQ -
page 92
3 2
Ln k
1 0
25 50 75 % actonitrile dans la phase mobile
Figure 6. Variations du Ln k en fonction du % dactonitrile dans la phase mobile pour le compos 3 Comment procderiez-vous pour obtenir un temps de rtention de 10 min pour le compos 3 ? Quelle est la perte de charge ?
Corrig n 9
Linvertase hydrolyse le saccharose en glucose et fructose. Le pic 3 qui disparat correspond donc au saccharose et les pics 2 et 3 qui augmentent au glucose et au fructose. Le jus dorange contient donc du glucose et du fructose en quantits identiques et du saccharose en quantit double comme le montrent les surfaces des pics et leurs variations (avec un coefficient de rponse identique pour les 3 oses). La rtention se fait par liaison hydrogne : chromatographie dadsorption. En effet tR diminue avec la masse molaire de lose et donc avec le nombre dOH susceptibles de donner une interaction hydrogne avec la phase stationnaire. Il est donc normal que les hexoses soient lus avec des tR infrieurs au diholoside quest la saccharose. Il pourrait sagir dune phase greffe polaire type NH2. Calcul du k pour le compos 3 qui est le saccharose : VM = t0 . D = . VT do
t0 =
tR est li au facteur de capacit par tR = t0 ( 1 + k) . Avec tR = 15 min pour le compos 3, k = 15,7
. VT 0,5 . 15 . 0,39 2 . = = 0,9 min D 4.1
En chromatographie dadsorption, k diminue avec laugmentation de o, cest dire avec la polarit de la phase mobile. Donc si le % dactonitrile diminue, le % deau augmente et la polarit aussi Si k diminue, ln k diminue et tR galement. Si k = 15,7 , Ln k = 2,75 et le graphe donne un % dactonitrile voisin de 75 %. Un tR de 10 min correspond un k de : 10 = 0,9 ( 1 + k) soit k = 10,1 et Ln k = 2,3. Par le graphe de la figure 6 il apparat alors quun pourcantage dactonitrile de 55 % dans la phase mobile permet dobtenir ce temps de rtention. La perte de charge est :
P =
h = 0,01 poise , L = 15 cm, v = D / s et s = s . v = 0,28 cm.sec-1 K0 = 0,07. 10-8 P = 60 bars
. L. v K0
Sujet n 10
Un broyat de graines de soja pralablement dlipid par pression est trait par de lhexane au moyen dun soxhlet. Le solvant est rcupr et soumis une vaporation sec sous vide partiel. Le rsidu est repris par un mlange compos dactonitrile, de mthanol et dacide sulfurique 0,5 % (50/25/25 , V/V/V). Une partie aliquote est injecte sur une colonne de 25 cm de longueur et 4,6 cm de diamtre interne dont la phase stationnaire est constitue de
r Jean-Louis CUQ -
page 93
particules sphriques de dodcyl- silice de 5 m de diamtre. La phase mobile, dont le dbit est de 1 ml.min-1, a la mme composition que celle du mlange ternaire dj dcrit. Les schmas des chromatogrammes talon et dosage sont donns sur la figure 1.
dosage
0 talon
3
2
6
3 4 5 6
9
7
12
8 9 10
minutes
12 minutes
figure 1 . Schmas des chromatogrammes de lextrait et de ltalon obtenu par injection de 10 l dune solution 10 M dacidesgras dans la phase mobile
2: 3: 4: 5: 6: acide linolnique acide palmitolique acide trans-hxadcnoque acide linolique acide trans-linolique (C18:3,cis) (C16:1,cis) (C16:1,trans) (C18:2,cis) (C18:2,trans) 7: 8: 9: 10: acide palmitique acide olique acide ladique acide starique (C16:O) (C18:1,cis) (C18:1,trans) (C18:O)
1 - Expliquer la dmarche analytique. Lajout dun talon interne serait-il judicieux ; quelle molcule choisiriez-vous, comment procderiez-vous et comment en tiendriez-vous compte dans la quantification. 2 - Quel est le principe de cette sparation ? Quel est le rle de lacide sulfurique dans la phase mobile ? 3 - Quel est le systme de dtection le mieux adapt ? 4 - La porosit interstitielle est gale 0,5. Quel est le facteur de capacit des composs majeurs spars partir de lextrait ? 5 - Comment peut-on agir sur les temps de rtention ? 6 - Quelle est la perte de charge, la viscosit de phase mobile est gale 1,2 cP 7 - Quelle est la rsolution entre les acides palmitique et olique ? 8 - Quelle est la slectivit de la colonne pour ces deux composs ?
Sujet n 11
Le dosage de lacide ascorbique et de quelques uns de ses drivs prsents dans une prparation base dorange est ralis de la faon suivante : 10 g de prparation sont mlangs 80 ml deau pralablement additionns avec prcision de 2,5 mg dacide propionique ; le broyat est alors complt 100 ml. Aprs centrifugation 5000 g pendant 10 min, le surnageant est pass sur une colonne de 5 cm de long et 1 cm de diamtre contenant des particules sphriques (dp = 10 m) de silice greffe C18. Lluat est rcupr et 100 l sont injects sur une colonne de 20 cm de longueur et 4,5 mm de diamtre contenant une phase stationnaire de type octadcyl silice dont les particules sphriques ont un diamtre moyen de 5 m. La phase mobile, dun dbit gal 1 ml.min-1 est compose dune solution aqueuse de bromure de ttrabutyl ammonium (0,2 %) et dun tampon phosphate 10 mM pH 6,5. Labsorbance de lluat est mesure 210 nm.
r Jean-Louis CUQ -
page 94
Le chromatogramme talon obtenu partir de solutions talons 0,025 g.l-1 et celui rsultant de lanalyse sont schmatiss sur la figure 1 (avec dans tous les cas une boucle externe de 100 l).
1 2 talon
3 dosage 4
1200
1000
740
1000
1000
10
tR (min)
15
320
10
350
tR (min)
15
figure 1 : chromatogrammes talon et dosage. ( 1 : acide ascorbique, 2 : acide dhydroascorbique, 3 : acide dictogulonique , 4 : acide propionique ). Les surfaces des pics sont indiques en valeurs relatives. 1) Expliquez le principe de cette sparation chromatographique. 2) Pourquoi passer le surnageant sur la colonne de 5 cm de longueur (C18) avant analyse ? 3) Sachant que la porosit de la colonne analytique de 20 cm de longueur est de 0,5, quelle est la valeur de to. 4) Dans ce systme, quels sont le facteur de capacit et la HEPT de lacide ascorbique. 5) La capacit de pics de la colonne dfinie par HERMAN est gale :
n=1+
avec N nombre de plateaux thoriques, t et t les temps de rtention du premier et dernier pic du chromatogramme. Comment exploiter cette donne ? 6) Comment diminueriez-vous les temps de rtention pour acclrer lanalyse ? 7) Quelle sont les concentrations en acides ascorbique, dhydroascorbique et dictogulonique de la prparation base dorange. Quel est le rle jou par lacide propionique ?
N . ln t 16 t
1) Sparation des acides organiques aprs appariement dions avec un ammonium apolaire. La paire dions forme apolaire est spare par chromatographie en phase inverse. Le pH est ajust une valeur suprieure au pKa de lacide et infrieure celui de lion dappariement.
Corrig n 11
HO HO
O H CH2 OH O H C
O H
COOH OH H C
OH Acide ascorbique Le pK1 de l'hydroxyle en C 3 est gal 4,04 25C
RCOOou RO+ N+ ((CH2)3 CH3)4
CH2 OH OH acide dictogulonique
RO- N+ ((CH2)3 CH3)4 paire dions apolaire
2) Le passage du surnageant sur C18 permet de retenir les composs apolaires qui pourraient gner dans la suite du dosage. 3) Graphiquement to est voisin de 1,5 min
370
r Jean-Louis CUQ -
page 95
Le volume de la colonne est gal Vt = L . r2 soit 20 . 3,14 . (0,45 / 2)2 = 3,18 cm3 VM = to.D = .Vt do to = .Vt / D = 0,5 . (3,18 / 1 ) = 1,6 min 4) tR = to ( 1 + k) Pour lacide ascorbique tR = 6 min do k = 2,75 Graphiquement on a = 1 min N = 16 ( tR / )2 soit N = 16 ( 6 / 1 )2 soit N = 576 HEPT = L / N do HEPT = 20 / 576 = 0,035 cm 5) n = 96 et les pics sont bien spars. Cet indice permet destimer la capacit dune colonne en terme de nombre de pics sparables avec une bonne rsolution. 6) La diminution des temps de rtention peut tre obtenue par une augmentation du dbit ou en diminuant le facteur de capacit en choisissant un ractif paire dions moins apolaire (par exemple un sel dammonium de type ttrapropyl ou ttramthyl). 7) Lacide propionique est un talon interne.Sa concentration initiale dans la prparation de fruit est de 2,5 mg / 100 ml. Ltalon analys directement a une concentration de 0,025 g.l-1 soit de 2,5 mg / 100 ml. On devrait donc obtenir sur les chromatogrammes talons et dosage la mme surface pour lacide propionique. Or pour ltalon on obtient une surface relative de 740 et pour le dosage de 370. Il faut donc corriger les rsultats par un facteur de 740 / 370 = 2. Pour lacide ascorbique : 0,025 . 2 = 0,050 g.l-1 de surnageant soit 5 mg / 100 ml ou 10 g de fruit Pour lacide dhydroascorbique : (0,025 . 2). 320 / 1000 = 1,6 mg / 100 ml ou 10 g de fruit Pour lacide dictogulonique : (0,025 . 2). 370 / 1200 = 1,54 mg / 100 ml ou 10 g de fruit
Sujet n 12
Une colonne chromatographique de 30 cm de longueur et 7,8 mm de diamtre interne contient une phase stationnaire constitue de sphres de polystyrne rticule avec du divinylbenzne; leur diamtre est de 5 m et leur porosit de 0,5. Elle est utilise avec une phase mobile compose dune solution aqueuse dacide phosphorique 0,05 N dont le dbit est de 0,5 ml.min-1. La temprature est de 30 C. Les analyses de 10 l dun mlange dacides organiques en solution 0,5 g pour 100 ml, ou dun extrait de pommes (20 g de pommes broys dans 80 ml deau et filtrs; on suppose que le volume final est de 100 ml) sont schmatises sur la figure 1.
Absorbance 210 nm 1 talon 2 3 4 extrait de pomme
10
15 tR (min)
10
15 tR (min)
Figure 1 : chromatogrammes talon et dosage. ( 1 : acide tartrique, 2 : acide malique, 3 : acide succinique , 4 : acide fumarique ). acide tartrique : COOH-CHOH-CHOH-COOH
r Jean-Louis CUQ -
page 96
acide malique : COOH-CH2-CHOH-COOH acide succinique : COOH-CH2-CH2-COOH acide fumarique : COOH-CH=CH-COOH 1) Expliquez le principe de cette sparation chromatographique. 2) Quelle est la valeur de to. 3) Quels sont le facteur de capacit et la HEPT de lacide malique. 4) Quel serait leffet dune augmentation de la temprature de la colonne ? 5) Comment diminueriez-vous les temps de rtention pour acclrer lanalyse ? 6) Quelle est la concentration en acide malique de la pomme ? 7) Quelles autres mthodes de dosage chromatographique sont-elles utilisables pour ce dosage ?
Sujet n 13
Pour analyser les acides organiques contenus dans du jus de raisin muscat on utilise une colonne chromatographique de 10 cm de longueur et 1 mm de diamtre interne. Elle contient une phase stationnaire constitue de sphres de polystyrne rticule avec du divinylbenzne; leur diamtre est de 1 m et leur porosit de 0,64. Elle est utilise avec une phase mobile compose dune solution aqueuse dacide phosphorique 0,06 N dont le dbit est de 25 L.min-1. La temprature est de 35 C. Les analyses de 5 L dun mlange dacides organiques en solution 0,4 g pour 100 mL, ou dun jus de raisin filtr sur membrane 0,2 m et pralablement additionn de 0,4 g dacide fumarique pour 100 mL sont schmatises sur la figure 1 (lacide fumarique est considr comme absent du jus de raisin).
Absorbance 210 nm
5 jus de raisin
S=1500
600
800
4 t (min) R
4 t (min) R
Figure 1 : chromatogrammes talon et dosage (1 : acide tartrique; 2 : acide ascorbique; 3 : acide malique ; 4 acide succinique ; 5 : acide fumarique). acide tartrique : COOH-CHOH-CHOH-COOH S = 1000 (exprime en unit arbitraire) acide ascorbique : CO-COH=COH-CH-CHOH-CH2OH S = 1200 O acide malique : COOH-CH2-CHOH-COOH S = 1050 acide succinique : COOH-CH2-CH2-COOH S = 1125 acide fumarique : COOH-CH=CH-COOH S = 975 1) Expliquez le principe de cette sparation chromatographique. 2) Quelle est la valeur de to. 3) Quels sont le facteur de capacit et la HEPT des acides tartrique et ascorbique. 4) Quel serait leffet dune augmentation de la temprature de la colonne ? 5) Comment diminueriez-vous les temps de rtention pour acclrer lanalyse ? 6) Quelle est la concentration en acide tartrique du jus ? Quel est le rle de lacide fumarique ? 7) Quelles autres mthodes de dosage chromatographique sont-elles utilisables pour ce dosage ?
Corrig n 13
r Jean-Louis CUQ -
page 97
1) Sparation des acides organiques par chromatographie en phase inverse. Le pH est ajust une valeur infrieure aux pKa des acides organiques pour viter leur ionisation et donc une trop forte polarit. Les acides sont spars en fonction de leur polarit : pour un mme nombre de carbone total (quatre par exemple) plus le nombre de fonctions OH est lev et plus le facteur de capacit est faible. 2) Graphiquement to est voisin de 2 min Le volume de la colonne est gal Vt = L . r2 soit 10 . 3,14 . (0,1 / 2)2 = 0,0785 cm3 VM = to.D = .Vt do to = .Vt / D = 0,64 . (0,0785 / 0,025 ) = 2 min 3) tR = to ( 1 + k) Pour lacide tartrique tR = 3,5 min do k = 0,75 Pour lacide ascorbique tR = 4 min do k = 1 Graphiquement on a 0,35 min pour les deux acides Pour lacide tartrique N = 16 (tR /)2 soit N = 16 (3,5 / 0,35)2 soit N = 1600 HEPT = L / N do HEPT = 10 / 1600 = 62,5 m Pour lacide ascorbique N = 16 (tR /)2 soit N = 16 (4 / 0,35)2 soit N = 2090 HEPT = L / N do HEPT = 10 / 2090 = 48 m 4) Une augmentation de la temprature augmentera k donc le temps de rtention. 5) La diminution des temps de rtention peut tre obtenue par une augmentation du dbit ou en diminuant le facteur de capacit (en abaissant le temprature ou en choisissant une phase mobile moins polaire (actonitrile ou mthanol en proportion adapte avec leau acidule). 6) Lacide fumarique constitue un talon interne. La concentration non corrige par cet talon interne est de :
concentration =
0,4 . S dosage 0,4. 1500 = 0,6 g. L-1 = S talon 1000
Lacide fumarique est la mme concentration dans ltalon et lchantillon dos. Or la surface du pic dosage montre quune perte est intervenue. La compensation de cette perte donne la concentration relle du jus de raisin :
concentration corrige =
0,4 . Sdosage S A fumarique talon 0,4. 1500 975 . = . = 0,73 g.L-1 Stalon Sac fumarique dosage 1000 800
7) Chromatographie dchange dions, par appariement dions ou encore CPG aprs drivatisation.
Vous aimerez peut-être aussi
- Parler Pour Que Les Enfants Ecoutent (PDFDrive)Document363 pagesParler Pour Que Les Enfants Ecoutent (PDFDrive)Eiluj Rag50% (2)
- Introduction à la physique de la matièreD'EverandIntroduction à la physique de la matièreÉvaluation : 3 sur 5 étoiles3/5 (1)
- Équilibres en solution: Les Grands Articles d'UniversalisD'EverandÉquilibres en solution: Les Grands Articles d'UniversalisPas encore d'évaluation
- Exercices Attribution RMN - 1D 2DDocument11 pagesExercices Attribution RMN - 1D 2DSabin Sam100% (1)
- Exercices Corrigés en ChromatographieDocument13 pagesExercices Corrigés en ChromatographiePeti' Pou89% (66)
- Glucides: Les Grands Articles d'UniversalisD'EverandGlucides: Les Grands Articles d'UniversalisPas encore d'évaluation
- Chromatographie Op PDFDocument41 pagesChromatographie Op PDFAbdelhakim BailalPas encore d'évaluation
- TD Chromatographie en Phase Gazeuse de L3 ProDocument6 pagesTD Chromatographie en Phase Gazeuse de L3 ProRicky Ranaivoson100% (5)
- Chromatographie: Les Grands Articles d'UniversalisD'EverandChromatographie: Les Grands Articles d'UniversalisPas encore d'évaluation
- Cours RMNDocument47 pagesCours RMNSanaa Laslami33% (3)
- Eléctrophorèse Capillaire THEORIE PDFDocument12 pagesEléctrophorèse Capillaire THEORIE PDFsalim100% (1)
- Microbiologie médicale II: stérilisation, diagnostic de laboratoire et réponse immunitaireD'EverandMicrobiologie médicale II: stérilisation, diagnostic de laboratoire et réponse immunitairePas encore d'évaluation
- Quelques Proprietes Atomiques Et Leurs Variations Dans La Classification PeriodiqueDocument57 pagesQuelques Proprietes Atomiques Et Leurs Variations Dans La Classification Periodiquearabe4ever100% (4)
- Les Méthodes ÉlectrochimiquesDocument83 pagesLes Méthodes ÉlectrochimiquesSalahbale100% (2)
- Exercices Corrigs de Spectroscopie Infrarouge PDFDocument2 pagesExercices Corrigs de Spectroscopie Infrarouge PDFJeffrey75% (16)
- Cours Spectroscopie de Masse PC3Document30 pagesCours Spectroscopie de Masse PC3Mahdi JmaiPas encore d'évaluation
- Cours CHAHER BAZIZI Nassima Méthodes D'analyses SpectroscopiquesDocument86 pagesCours CHAHER BAZIZI Nassima Méthodes D'analyses SpectroscopiquesimmPas encore d'évaluation
- Alexandra Henrion 130829Document1 pageAlexandra Henrion 130829Nathalie de L'amfePas encore d'évaluation
- Analyse Qualitative Et Quantitative s3Document24 pagesAnalyse Qualitative Et Quantitative s3safemind67% (3)
- Cours Spectrométrie Masse.3eme Année Chimie OrganiqueDocument64 pagesCours Spectrométrie Masse.3eme Année Chimie OrganiqueGüzel GözlerPas encore d'évaluation
- TD Chromatographie CPG (Y.françois)Document9 pagesTD Chromatographie CPG (Y.françois)FATIMA KEFIFPas encore d'évaluation
- Serie4 ExosDocument4 pagesSerie4 Exossalima sousou50% (4)
- C514 TD Serie 5 CORRECTION - 2020-2021Document6 pagesC514 TD Serie 5 CORRECTION - 2020-2021simo Gri100% (3)
- Centre de Formation Grandir Autrement, Catalogue Des ServicesDocument34 pagesCentre de Formation Grandir Autrement, Catalogue Des ServicesBenoit CloutierPas encore d'évaluation
- Corrigés Exercices - Spectres RMNDocument16 pagesCorrigés Exercices - Spectres RMNsana sanaPas encore d'évaluation
- HPLCDocument46 pagesHPLCDjoe LalaounaPas encore d'évaluation
- Série 4 Chromato - QualitativeDocument3 pagesSérie 4 Chromato - QualitativeMohamed Dahmane100% (2)
- CPM701 Exo RMNDocument13 pagesCPM701 Exo RMNDomingos Morais ManuelPas encore d'évaluation
- ChapM1 Chimie GénéraleDocument99 pagesChapM1 Chimie GénéraleHakim Bil100% (2)
- Chromatographie Master1Document30 pagesChromatographie Master1nabil86% (7)
- TP - Spectroscopie IR Et RMN - Correction-ConvertiDocument5 pagesTP - Spectroscopie IR Et RMN - Correction-ConvertiRania SahbiPas encore d'évaluation
- Serie7 Exos PDFDocument2 pagesSerie7 Exos PDFsalima sousouPas encore d'évaluation
- Corrige Exercices RMN ProtonsDocument2 pagesCorrige Exercices RMN ProtonsYean-baptiste Dupas Decallais83% (6)
- BadulescuDocument13 pagesBadulescuBassem EllafiPas encore d'évaluation
- APPLICATION ChromatographieDocument2 pagesAPPLICATION ChromatographieNino Nini Nino0% (1)
- TD Methodes Danalyse 19 20Document15 pagesTD Methodes Danalyse 19 20stani kamdoum0% (1)
- Chromatographie Sur Couche Mince 2010Document1 pageChromatographie Sur Couche Mince 2010LeifEricssonPas encore d'évaluation
- L3 TAA Toutes Les Series TD TABCorrigésDocument27 pagesL3 TAA Toutes Les Series TD TABCorrigésIbtissam HmPas encore d'évaluation
- Serie1 CorrectionsDocument4 pagesSerie1 CorrectionsSaber Ben Zian100% (1)
- Chromatographie NotionsDocument55 pagesChromatographie NotionsJavier Andres Gonzalez Prieto100% (1)
- RMN (2017-2018)Document58 pagesRMN (2017-2018)Walid AbouloifaPas encore d'évaluation
- CC2 17 enDocument6 pagesCC2 17 enWinna HaPas encore d'évaluation
- Chromato2 Phase LiquideDocument82 pagesChromato2 Phase Liquideraftoupin100% (1)
- Cours de Spectrometrie de Masse 1Document16 pagesCours de Spectrometrie de Masse 1Reda Rami50% (2)
- TP 3Document3 pagesTP 3Ahmed YounsiPas encore d'évaluation
- Chromatographie en Phase LiquideDocument2 pagesChromatographie en Phase LiquidePaul Fathead100% (1)
- Extractions Et ChromatographiesDocument1 pageExtractions Et ChromatographiesAbderrahime Chikhaoui100% (7)
- RMN 2DDocument14 pagesRMN 2Dezo ezoPas encore d'évaluation
- Chromatographie en Phase Gazeuse: EXERCICE 4.1. On Souhaite Séparer Un Mélange de N-Heptane (TDocument4 pagesChromatographie en Phase Gazeuse: EXERCICE 4.1. On Souhaite Séparer Un Mélange de N-Heptane (TWinna Ha100% (2)
- Cour HPLCDocument36 pagesCour HPLCSanaPas encore d'évaluation
- Cour Exercice SpectroscopieDocument67 pagesCour Exercice SpectroscopieChk YahyaPas encore d'évaluation
- HPLCDocument45 pagesHPLCMohamed Taieb Bakouche100% (2)
- CPGDocument4 pagesCPGNadia HassinaPas encore d'évaluation
- ChromatographieDocument10 pagesChromatographieAbdelhakim BailalPas encore d'évaluation
- TD Chromatographie en Phase Gazeuse de L3 CorrectionDocument7 pagesTD Chromatographie en Phase Gazeuse de L3 CorrectionChenini Tayeb100% (1)
- TD 5 - 2020-21Document2 pagesTD 5 - 2020-21lahyani100% (1)
- 2.spectres Infrarouge PDFDocument6 pages2.spectres Infrarouge PDFkimmik100% (1)
- LPro Chromato PDFDocument81 pagesLPro Chromato PDFanon_509931202Pas encore d'évaluation
- Titrage AmpérométriqueDocument13 pagesTitrage AmpérométriqueSellam AnisPas encore d'évaluation
- Exm Atg Master Oxalate de Ca Gypse 2015 2019 PDFDocument1 pageExm Atg Master Oxalate de Ca Gypse 2015 2019 PDFVerginica Spilosoma100% (1)
- Cours Chromatograpie 2 3 EtudiantsOIDDocument88 pagesCours Chromatograpie 2 3 EtudiantsOIDRAZIQ YOUSSEFPas encore d'évaluation
- Applications de la spectrophotomérie en phytochimie: sciencesD'EverandApplications de la spectrophotomérie en phytochimie: sciencesPas encore d'évaluation
- Faculté Pluridisciplinaire de Nador AnnéeDocument3 pagesFaculté Pluridisciplinaire de Nador AnnéeHakim BilPas encore d'évaluation
- 3.ato - 04 AtomistiqueDocument3 pages3.ato - 04 AtomistiqueHakim BilPas encore d'évaluation
- Session D'examens: Date Examen ModuleDocument3 pagesSession D'examens: Date Examen ModuleHakim BilPas encore d'évaluation
- Corrigé Dissertation Type BaxDocument3 pagesCorrigé Dissertation Type Baxonedesco89Pas encore d'évaluation
- Les Echos 28 Février Solaire Ou Biosourcé, Le Ciment Zéro Carbone Se ProfileDocument2 pagesLes Echos 28 Février Solaire Ou Biosourcé, Le Ciment Zéro Carbone Se ProfilepaulPas encore d'évaluation
- Petra KantorDocument3 pagesPetra KantorZvonirPas encore d'évaluation
- Teigne - Quel Traitement ? - Fiches Santé Et Conseils MédicauxDocument2 pagesTeigne - Quel Traitement ? - Fiches Santé Et Conseils MédicauxNaruto & SasukePas encore d'évaluation
- Programme D'études TSC-AGC-TSC 2007Document92 pagesProgramme D'études TSC-AGC-TSC 2007Finance100% (1)
- GRH 154 0087Document4 pagesGRH 154 0087Martin DansouPas encore d'évaluation
- Guide Print PDFDocument181 pagesGuide Print PDFisomoaPas encore d'évaluation
- Quotidien N°3571 - 0Document42 pagesQuotidien N°3571 - 0IbrahimPas encore d'évaluation
- SlidesCours Part1Document22 pagesSlidesCours Part1ayoubPas encore d'évaluation
- Theme Onde SM SujetDocument21 pagesTheme Onde SM SujetAlphaPas encore d'évaluation
- CHAP 2 (L'énergie Chimique)Document4 pagesCHAP 2 (L'énergie Chimique)hepived840Pas encore d'évaluation
- Introduction À La Publicité DisplayDocument7 pagesIntroduction À La Publicité Displaydjado nitorPas encore d'évaluation
- Les Parties A Et B Sont IndépendantesDocument6 pagesLes Parties A Et B Sont IndépendantesSaraPas encore d'évaluation
- BudgétisationDocument9 pagesBudgétisationSaad JamaaPas encore d'évaluation
- Corrige Type Interogation Semestre Ingenieurie HumaineDocument2 pagesCorrige Type Interogation Semestre Ingenieurie Humainejihadraw92Pas encore d'évaluation
- Chapitre 9 ST CorrDocument2 pagesChapitre 9 ST Corr6233814Pas encore d'évaluation
- TFC - MAKUBAMA Sur La Perception Et La Hiérarchisation Des Banques Par Les Clients de Mbanza Ngungu. Cas de La RAWBANK, EQUITY BCDC Et La FIRST BANKDocument43 pagesTFC - MAKUBAMA Sur La Perception Et La Hiérarchisation Des Banques Par Les Clients de Mbanza Ngungu. Cas de La RAWBANK, EQUITY BCDC Et La FIRST BANKPirate Kopp45Pas encore d'évaluation
- 2004 Chandon Echelle Ordinale Permettant de Clqsser Les Répondants en SatisfaitDocument15 pages2004 Chandon Echelle Ordinale Permettant de Clqsser Les Répondants en SatisfaitnadjahPas encore d'évaluation
- SC La Revelation Devoir de Conges Année Scolaire 2023 - 2024Document14 pagesSC La Revelation Devoir de Conges Année Scolaire 2023 - 2024John LocationPas encore d'évaluation
- Roland PM 30 Manuel Utilisateur FR 28981Document16 pagesRoland PM 30 Manuel Utilisateur FR 28981AlcorgoldoPas encore d'évaluation
- 8-Etude Du Comportement Mecanique Des Chaussees SoupDocument7 pages8-Etude Du Comportement Mecanique Des Chaussees SoupWissem TaktakPas encore d'évaluation
- Devis JABALI 2110Document1 pageDevis JABALI 2110Abdo MoghéètPas encore d'évaluation
- La Structure Staff and LineDocument13 pagesLa Structure Staff and LinedohaPas encore d'évaluation
- Sujet 2Document6 pagesSujet 2Eba Jean aymardPas encore d'évaluation
- Rapport de Diagnostique de L'escalatorDocument3 pagesRapport de Diagnostique de L'escalatorMichael WendeuPas encore d'évaluation
- Spatial Temporal Au Min App-FDocument24 pagesSpatial Temporal Au Min App-FKeurth Kassa KassaPas encore d'évaluation