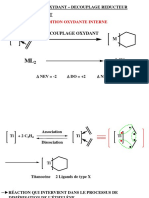
Vous aimerez peut-être aussi
- Matérialisation AutomatismeDocument41 pagesMatérialisation AutomatismeGuillaume NchupassePas encore d'évaluation
- 2009 Lyon Walchshofer Radix StrucMolOrgaCours PDFDocument19 pages2009 Lyon Walchshofer Radix StrucMolOrgaCours PDFAbdessamed GherbaouiPas encore d'évaluation
- 15 - TD - Procédures - Doc ERMDocument9 pages15 - TD - Procédures - Doc ERMjugoPas encore d'évaluation
- TD Flexion Et Structure Metalliques Correction PDFDocument5 pagesTD Flexion Et Structure Metalliques Correction PDFchaudronnierPas encore d'évaluation
- DP OSmath-phys Corrigé 10VPDocument9 pagesDP OSmath-phys Corrigé 10VPEvis BashaPas encore d'évaluation
- Aldehydes Et CetonesDocument24 pagesAldehydes Et CetonesAhmed Ben AmmarPas encore d'évaluation
- Reactivité Chimique - Chapitre 1Document44 pagesReactivité Chimique - Chapitre 1abraham LincolnPas encore d'évaluation
- SERIE 4 CorrigéDocument8 pagesSERIE 4 CorrigéMouflehPas encore d'évaluation
- Chim 306 - Notes de CoursDocument43 pagesChim 306 - Notes de Coursnatural recipePas encore d'évaluation
- Cours Introduction Chim I e Compos ÉsDocument73 pagesCours Introduction Chim I e Compos Ésأمين حسونيPas encore d'évaluation
- CH - Coord SMC6 2021 - 2022 AamiliDocument42 pagesCH - Coord SMC6 2021 - 2022 AamiliNabil mifdalPas encore d'évaluation
- Bilan TPO - 02Document12 pagesBilan TPO - 02Simona StefanPas encore d'évaluation
- Chap IIIDocument23 pagesChap IIIfadoua lakouissiPas encore d'évaluation
- Les Grands Classe de La Chimie OrganiqueDocument7 pagesLes Grands Classe de La Chimie Organiqueayyoub dhbPas encore d'évaluation
- Techniques VolumetriquesDocument33 pagesTechniques VolumetriquesAoulmi AmaniPas encore d'évaluation
- Série 5 Et CorrigéDocument7 pagesSérie 5 Et CorrigéChoaib NouhyPas encore d'évaluation
- Chap 4Document13 pagesChap 4Chihab GhlalaPas encore d'évaluation
- Chimie Organique Générale Cours 03 Universite Ibn Tofail KenitraDocument69 pagesChimie Organique Générale Cours 03 Universite Ibn Tofail KenitraJamal KOUBACHIPas encore d'évaluation
- Catalyse HeterogeneDocument132 pagesCatalyse HeterogenelilyaPas encore d'évaluation
- U4orgaanas1 00cDocument2 pagesU4orgaanas1 00cEssamiPas encore d'évaluation
- Chapitre IDocument25 pagesChapitre IKurtusPas encore d'évaluation
- Cours Chimie Organique 2Document11 pagesCours Chimie Organique 2gregoire korgaPas encore d'évaluation
- TD Orbitales MoléculairesDocument6 pagesTD Orbitales MoléculairesChimiste Chimiste0% (2)
- Biomatériaux À Base Ce CelluloseDocument132 pagesBiomatériaux À Base Ce CelluloseEL Hafedh EL MouhabPas encore d'évaluation
- Chim430 Organometallique Poly de Cours 2012 2013Document133 pagesChim430 Organometallique Poly de Cours 2012 2013AchrafAssil100% (5)
- Id-3031 PDFDocument29 pagesId-3031 PDFMicrocosmos MystiquePas encore d'évaluation
- 2.sélectivité en Chimie Organique PDFDocument4 pages2.sélectivité en Chimie Organique PDFkimmikPas encore d'évaluation
- Master SiliciumDocument56 pagesMaster SiliciumMohamed EL FAGHLOUMIPas encore d'évaluation
- TD Nomenclature Corrigé12goodprepa PDFDocument3 pagesTD Nomenclature Corrigé12goodprepa PDFNour EddinePas encore d'évaluation
- Chapitre 4 Les Réactions Doxydo ReductionDocument57 pagesChapitre 4 Les Réactions Doxydo ReductionabdelouahedPas encore d'évaluation
- Cours 1 de Chimie Des Solutions Chapitre 1 ConductimétrieDocument47 pagesCours 1 de Chimie Des Solutions Chapitre 1 ConductimétrieRaouia MakhloufPas encore d'évaluation
- Examen TP Coordination 2021-2022Document1 pageExamen TP Coordination 2021-2022Mohamed TaouilPas encore d'évaluation
- Orga 6Document11 pagesOrga 6Romain Laher100% (1)
- Rétrosynthèse, Corrigés Des ExercicesDocument19 pagesRétrosynthèse, Corrigés Des Exercicesfantamat974Pas encore d'évaluation
- Cours Catalyse Heterogene 2020 DaloaDocument42 pagesCours Catalyse Heterogene 2020 DaloaMohamed CoulibalyPas encore d'évaluation
- Rof1 - TD 2022Document10 pagesRof1 - TD 2022Lisa CrpsPas encore d'évaluation
- 4 Orga 2 PDFDocument80 pages4 Orga 2 PDFKamel HachamaPas encore d'évaluation
- Vissac Argiles Biopolymeres PDFDocument80 pagesVissac Argiles Biopolymeres PDFetopec7440Pas encore d'évaluation
- ABDESSELAM FatimaZohraDocument51 pagesABDESSELAM FatimaZohraNoura AzaouagPas encore d'évaluation
- Chapitre 1 CorrigéDocument11 pagesChapitre 1 CorrigéYoussef ArbaPas encore d'évaluation
- 011 PDFDocument88 pages011 PDFlolobsPas encore d'évaluation
- Liste de Réactions en Chimie Organique PCSIDocument3 pagesListe de Réactions en Chimie Organique PCSIlebananistPas encore d'évaluation
- ElectrochimieDocument30 pagesElectrochimieawara237Pas encore d'évaluation
- DiolDocument7 pagesDiolFrederic WustPas encore d'évaluation
- Affiche Master PharmacoDocument1 pageAffiche Master PharmacobnhikPas encore d'évaluation
- Molécule FuraneDocument11 pagesMolécule FuraneHamza BoulikaPas encore d'évaluation
- Organul 1Document17 pagesOrganul 1Wiwi Laura100% (1)
- TD5 PharmacieDocument2 pagesTD5 PharmacieAycha MedPas encore d'évaluation
- Cours 5 Nomenclature SNV L1 2020 2021Document30 pagesCours 5 Nomenclature SNV L1 2020 2021Mohamed DjaniPas encore d'évaluation
- Exercices Tensioactifsemulsions MoussesDocument3 pagesExercices Tensioactifsemulsions MoussesYassine RakchoPas encore d'évaluation
- Cours Chimie IIDocument37 pagesCours Chimie IINesrine Kaddouri100% (1)
- Chapitre 2-Méthodes D'obtention Des Molécules Optiquement ActivesDocument26 pagesChapitre 2-Méthodes D'obtention Des Molécules Optiquement ActivesMériem BradaiPas encore d'évaluation
- TD26 Diagramme D'ellingham Du Mercure-CorrigeDocument2 pagesTD26 Diagramme D'ellingham Du Mercure-Corrigehanane hanounePas encore d'évaluation
- Cours4 Les Reactions Chimie Organique Part1Document29 pagesCours4 Les Reactions Chimie Organique Part1Kalosoiretrotchgmail.com KalosoPas encore d'évaluation
- Catalyseur de WilkinsonDocument37 pagesCatalyseur de WilkinsonAchwak BelfadelPas encore d'évaluation
- QQQ PDFDocument62 pagesQQQ PDFSalma DaaddiPas encore d'évaluation
- TD Extraction Des Substances NaturellesDocument3 pagesTD Extraction Des Substances Naturellesnassima chenikhaPas encore d'évaluation
- Travaux Dirigés Sur Les Polymères Et Composites ConvertiDocument16 pagesTravaux Dirigés Sur Les Polymères Et Composites ConvertiAy Ou ChaPas encore d'évaluation
- Examen de Chimie MinéraleDocument7 pagesExamen de Chimie Minéraleranaater008Pas encore d'évaluation
- Exercices de Nomenclature-2Document9 pagesExercices de Nomenclature-2Oussama ChemroukPas encore d'évaluation
- Chap II ComplexationDocument17 pagesChap II ComplexationyoussefPas encore d'évaluation
- Applications de la spectrophotomérie en phytochimie: sciencesD'EverandApplications de la spectrophotomérie en phytochimie: sciencesPas encore d'évaluation
- Aldehydes Ce TonesDocument31 pagesAldehydes Ce Tonesmariem outahmiditPas encore d'évaluation
- Thème: Chimie Organique: Niveau: T CDE DisciplineDocument9 pagesThème: Chimie Organique: Niveau: T CDE DisciplineXrap HitPas encore d'évaluation
- Beni Mellal - PC - FR - RDocument1 pageBeni Mellal - PC - FR - RSalah LáálámPas encore d'évaluation
- Cour Potentiel Standardd'electrodeDocument2 pagesCour Potentiel Standardd'electrodeSalah LáálámPas encore d'évaluation
- raw:/storage/emulated/0/Download/Browser/Annonce Rentree Universitaire 2020 2021Document1 pageraw:/storage/emulated/0/Download/Browser/Annonce Rentree Universitaire 2020 2021Salah LáálámPas encore d'évaluation
- raw:/storage/emulated/0/Download/Browser/COURS-TD CATALYSE Partie IIDocument15 pagesraw:/storage/emulated/0/Download/Browser/COURS-TD CATALYSE Partie IISalah LáálámPas encore d'évaluation
- raw:/storage/emulated/0/Download/Browser/fiche LPCDocument2 pagesraw:/storage/emulated/0/Download/Browser/fiche LPCSalah LáálámPas encore d'évaluation
- Letexte04 PDFDocument4 pagesLetexte04 PDFSalah LáálámPas encore d'évaluation
- Chapitre IDocument8 pagesChapitre ISalah LáálámPas encore d'évaluation
- Huckel PDFDocument13 pagesHuckel PDFSalah LáálámPas encore d'évaluation
- Absorption DeauDocument4 pagesAbsorption DeauChemali RamziPas encore d'évaluation
- Correction Évaluation N° 3 2022-2023Document2 pagesCorrection Évaluation N° 3 2022-2023popi22377Pas encore d'évaluation
- Exercices CorrigésDocument5 pagesExercices CorrigésAboubakar Nambiema100% (5)
- Chapitre 3 - Caract ®risation Physico-Chimique, Structurale Et M ®canique Des Mat ®riauxDocument87 pagesChapitre 3 - Caract ®risation Physico-Chimique, Structurale Et M ®canique Des Mat ®riauxSilouane HubertPas encore d'évaluation
- OnduleurDocument11 pagesOnduleurELmokhtar HamrouniPas encore d'évaluation
- Les Éléments Chimiques Dans L'univers - 1ère - Cours Enseignement Scientifique - KartableDocument2 pagesLes Éléments Chimiques Dans L'univers - 1ère - Cours Enseignement Scientifique - KartableHello HelloPas encore d'évaluation
- Présentation Synthése Rapport FD9 JNAIKHDocument21 pagesPrésentation Synthése Rapport FD9 JNAIKHrfaliPas encore d'évaluation
- TEXUM Solutions TexgripDocument2 pagesTEXUM Solutions TexgripLo MPas encore d'évaluation
- TP1 GR4 2GC3Document7 pagesTP1 GR4 2GC3Anas RachydPas encore d'évaluation
- TDelectrocinetiqueCh2v1 0Document8 pagesTDelectrocinetiqueCh2v1 0Faiçal Saidi100% (2)
- Poteau Recommandations Prof EC2Document8 pagesPoteau Recommandations Prof EC2Jean-Vincent KandelaPas encore d'évaluation
- TP N°2Document1 pageTP N°2Marouane TaibiniPas encore d'évaluation
- Cuivre DRV ToshibaDocument181 pagesCuivre DRV ToshibaOULDITTOU Mohamed100% (2)
- Béton FinalDocument140 pagesBéton FinalRith MakaraPas encore d'évaluation
- ForgeageDocument26 pagesForgeageThomas Barraud50% (2)
- Exo 22 EvDocument4 pagesExo 22 EvZakariaElkatibPas encore d'évaluation
- ECLAIRAGE AvionDocument18 pagesECLAIRAGE AvionHelmi ghorbelPas encore d'évaluation
- Cours de Systemes AsservisDocument82 pagesCours de Systemes Asserviselectroblida86% (7)
- CPHY423 TD TKleinDocument11 pagesCPHY423 TD TKleinChaabani WajdiPas encore d'évaluation
- SommaireDocument40 pagesSommaireNajoua El Housni100% (1)
- Physica Magazine Avril 2018Document31 pagesPhysica Magazine Avril 2018Bouchra Hiba OurahmounePas encore d'évaluation
- 3.ato - 04 AtomistiqueDocument3 pages3.ato - 04 AtomistiqueHakim BilPas encore d'évaluation
- M1101 - DS Vé Réglable II - v2014 PDFDocument8 pagesM1101 - DS Vé Réglable II - v2014 PDFRepesse SebastienPas encore d'évaluation
- Debitmetre AnnubarDocument22 pagesDebitmetre AnnubarBAMBA Lango RichardPas encore d'évaluation
- Métrologie Dimensionnelle TPDocument24 pagesMétrologie Dimensionnelle TPOumar Sall100% (8)
- PDF ElectriciteDocument5 pagesPDF Electriciteʚḯɞ Mäḑjïdoü Bãrcelønē ʚḯɞPas encore d'évaluation