Vous aimerez peut-être aussi
- Seminaire-DALI 2013 (Enregistré Automatiquement)Document26 pagesSeminaire-DALI 2013 (Enregistré Automatiquement)nini zerdjebPas encore d'évaluation
- Cours Chimie Organique FS Ben M'Sik 2012-2013Document47 pagesCours Chimie Organique FS Ben M'Sik 2012-2013Mahfoud ZakiPas encore d'évaluation
- 502-JMES-2363-Ben MoussaDocument13 pages502-JMES-2363-Ben MoussaMalek abidiPas encore d'évaluation
- Bio MateriauxDocument6 pagesBio MateriauxHoussam SaadPas encore d'évaluation
- EVA 2-3 Métabolisme Des Microorganismes - 2Document5 pagesEVA 2-3 Métabolisme Des Microorganismes - 2Kenza EL KortbiPas encore d'évaluation
- Chapitre I-Synthese BibliographiqueDocument39 pagesChapitre I-Synthese BibliographiqueGabriel BenyahiaPas encore d'évaluation
- PDF Translator 1599129695172 PDFDocument74 pagesPDF Translator 1599129695172 PDFPalmire TshakoPas encore d'évaluation
- Fottation Minerais ChimIndusDocument54 pagesFottation Minerais ChimIndusoulamine hichamPas encore d'évaluation
- Bio 12ème SE-1Document30 pagesBio 12ème SE-1Jœël Crèspœ KhæmænœPas encore d'évaluation
- Chapitre 1 Principaux Adsorbants IndustrielsDocument34 pagesChapitre 1 Principaux Adsorbants IndustrielsElaziouti AbdelkaderPas encore d'évaluation
- As 9 (1) (2013) 3Document11 pagesAs 9 (1) (2013) 3EL Hassania EL HERRADIPas encore d'évaluation
- EcorrectionXAMEN MDD master 2 en 24 (1)Document3 pagesEcorrectionXAMEN MDD master 2 en 24 (1)kadere83Pas encore d'évaluation
- 28alcalino TerreuxDocument10 pages28alcalino Terreuxalaae medPas encore d'évaluation
- BBA - Chapitre 1 Et 2 Chimie Organique - Bahloul Ahmed - L2Document16 pagesBBA - Chapitre 1 Et 2 Chimie Organique - Bahloul Ahmed - L2Imene Aoun SeghirPas encore d'évaluation
- Physiologie Vegetale Cours 5photosynthese2Document12 pagesPhysiologie Vegetale Cours 5photosynthese2SALAH-EDDINE AZIZIPas encore d'évaluation
- Chimie OrganométalliqueDocument2 pagesChimie OrganométalliqueMePas encore d'évaluation
- Chim430 Organometallique Cours PDFDocument144 pagesChim430 Organometallique Cours PDFAmel AyadiPas encore d'évaluation
- Chimie Organique EnamDocument68 pagesChimie Organique Enammontassirhalouane5Pas encore d'évaluation
- TP 4 LezoulDocument11 pagesTP 4 Lezoulcsdfs.chimiePas encore d'évaluation
- Cours Chimie Organique FSF 2007-2008Document52 pagesCours Chimie Organique FSF 2007-2008Mahfoud ZakiPas encore d'évaluation
- TD Chimie Minerale 2024Document2 pagesTD Chimie Minerale 2024muslim76663150Pas encore d'évaluation
- Matériaux PhosphatésDocument61 pagesMatériaux Phosphatésmerati100% (1)
- Master SiliciumDocument56 pagesMaster SiliciumMohamed EL FAGHLOUMIPas encore d'évaluation
- Rapport de stage Axel Krzyzaniak M1 ICMDocument36 pagesRapport de stage Axel Krzyzaniak M1 ICMhalimaelbouamiPas encore d'évaluation
- Généralités Sur Le PhosphoreDocument6 pagesGénéralités Sur Le Phosphorejawad azPas encore d'évaluation
- Matériaux Inorganiques PoreuxDocument35 pagesMatériaux Inorganiques PoreuxAna-Marija Bartolincic100% (1)
- Chapitre IDocument27 pagesChapitre IMoujahed Farés100% (1)
- Problématique?: Colorants Pollution EnvironnementDocument47 pagesProblématique?: Colorants Pollution EnvironnementBouchra NasriPas encore d'évaluation
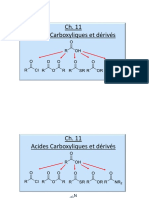
- Ch11 Acides Esters AmidesDocument56 pagesCh11 Acides Esters AmidesYugi kevin14Pas encore d'évaluation
- Organique Chapitre1Document9 pagesOrganique Chapitre1tsikpawoclementPas encore d'évaluation
- Caractérisation Des Eponges Version FinalDocument25 pagesCaractérisation Des Eponges Version FinalwidadPas encore d'évaluation
- Oxalatation Des Aciers SpéciauxDocument5 pagesOxalatation Des Aciers SpéciauxOualidPas encore d'évaluation
- Diagramme Fer CarboneDocument30 pagesDiagramme Fer Carbonedayang100% (1)
- Biochimie Approfondie Cours 8Document59 pagesBiochimie Approfondie Cours 8Alhassane BahPas encore d'évaluation
- Chimie Mineral 1Document9 pagesChimie Mineral 1Christy KlPas encore d'évaluation
- 1 PBDocument23 pages1 PBBrou Guillaume KOUASSIPas encore d'évaluation
- Biochimie 1Document26 pagesBiochimie 1Omar Makhlouf100% (1)
- Cour MinéralogieDocument14 pagesCour MinéralogieabdouPas encore d'évaluation
- Chap6 AlcanesDocument57 pagesChap6 AlcanesJeff WarrenPas encore d'évaluation
- Asgb 93 81Document6 pagesAsgb 93 81Bonheur NumbiPas encore d'évaluation
- CHO Phar 2Document79 pagesCHO Phar 2BADRXCKPas encore d'évaluation
- SilicatesDocument20 pagesSilicatesbokkun100% (1)
- Microbiolo S6Document25 pagesMicrobiolo S6Mlle BushaPas encore d'évaluation
- 2 G N Ralit S - B.LEBEAUDocument32 pages2 G N Ralit S - B.LEBEAUMourad MakhloufPas encore d'évaluation
- La Minéralogie 2Document15 pagesLa Minéralogie 2Nada serine ZouanePas encore d'évaluation
- TPN 13: Diagrammes Potentiel-Ph, Oxydoréduction, Constantes ThermodynamiquesDocument21 pagesTPN 13: Diagrammes Potentiel-Ph, Oxydoréduction, Constantes Thermodynamiqueszazaziri201Pas encore d'évaluation
- FALLET 2015 Archivage CorDocument166 pagesFALLET 2015 Archivage CorAbdoulaye HaouaPas encore d'évaluation
- 2021 Echelles Du VivantDocument18 pages2021 Echelles Du VivantAbubakr SalhiPas encore d'évaluation
- Cours de Cristallographie Et MineralogieDocument47 pagesCours de Cristallographie Et MineralogieIB AlayiPas encore d'évaluation
- Terminologie MetauxDocument20 pagesTerminologie MetauxMourad RabahPas encore d'évaluation
- Echanges D Ions Chromatographie Ionique Et Mise en Oeuvre Industrielle Des Resines SommaireDocument10 pagesEchanges D Ions Chromatographie Ionique Et Mise en Oeuvre Industrielle Des Resines SommaireMohamed El Mehdi MEKHZOUMPas encore d'évaluation
- TP N°18 - Corrosion Et Protection Du FerDocument11 pagesTP N°18 - Corrosion Et Protection Du FerLou BalonPas encore d'évaluation
- Cours BIOMATERIAUX 2019 20 Converti (1) 2Document34 pagesCours BIOMATERIAUX 2019 20 Converti (1) 2Soukaina Laghdass100% (1)
- Organo PhosphoréDocument11 pagesOrgano PhosphoréZied BouabenePas encore d'évaluation
- 10 Nitration D Un Noyau Aromatique 1Document4 pages10 Nitration D Un Noyau Aromatique 1Oussama IdrissPas encore d'évaluation
- ChimieMinSyst IRDDocument36 pagesChimieMinSyst IRDKalosoiretrotchgmail.com KalosoPas encore d'évaluation
- Technologie de l’acier: Les Grands Articles d'UniversalisD'EverandTechnologie de l’acier: Les Grands Articles d'UniversalisPas encore d'évaluation
- Le cuivre et le plomb dans l'alimentation et l'industrie, au point de vue de l'hygièneD'EverandLe cuivre et le plomb dans l'alimentation et l'industrie, au point de vue de l'hygiènePas encore d'évaluation
- La Pierre Philosophale: Grand éclaircissement et Preuves irréfutables de son existenceD'EverandLa Pierre Philosophale: Grand éclaircissement et Preuves irréfutables de son existenceÉvaluation : 4 sur 5 étoiles4/5 (3)
- Photosynthèse: Les Grands Articles d'UniversalisD'EverandPhotosynthèse: Les Grands Articles d'UniversalisPas encore d'évaluation
- XRD Université de MontréalDocument15 pagesXRD Université de MontréalTrà NguyễnPas encore d'évaluation
- Radiocristallographie-1Document14 pagesRadiocristallographie-1dibrawan18Pas encore d'évaluation
- CH2-La Radio-Cristallographie-EtudiantsDocument26 pagesCH2-La Radio-Cristallographie-EtudiantsMr. HansPas encore d'évaluation
- Cours Diffraction Des RayonsX Sur PoudresDocument15 pagesCours Diffraction Des RayonsX Sur PoudresEmma StonePas encore d'évaluation
- These IaicheDocument233 pagesThese Iaicheasma soumaPas encore d'évaluation
- Rapport de Visite de Plateau Technique D'analyses Physico-Chimiques USTHBDocument23 pagesRapport de Visite de Plateau Technique D'analyses Physico-Chimiques USTHBEskander SamPas encore d'évaluation
- Bouraada CourDocument11 pagesBouraada CourBerzigue BelabbesPas encore d'évaluation
- Prévision Et Construction Des Diagrammes de Phases Des Systèmes Métalliques Ternaires À Partir Des Binaires Limitrophes PDFDocument98 pagesPrévision Et Construction Des Diagrammes de Phases Des Systèmes Métalliques Ternaires À Partir Des Binaires Limitrophes PDFBelgasem Assel100% (1)
- Cours RXDocument36 pagesCours RXZerPas encore d'évaluation
- Chap 15 ADocument15 pagesChap 15 ARachid MakhloufiPas encore d'évaluation
- 03 - Structure Cristalline Des Solides (Mode de Compatibilité)Document53 pages03 - Structure Cristalline Des Solides (Mode de Compatibilité)Imane NahPas encore d'évaluation
- TexteDocument9 pagesTextetracy massomaPas encore d'évaluation
- RX StructureDocument15 pagesRX StructureSIAKA OuattaraPas encore d'évaluation
- ChapIV RadioDocument21 pagesChapIV RadioHananePas encore d'évaluation
- Comment Rédiger Un Rapport ScientifiqueDocument15 pagesComment Rédiger Un Rapport ScientifiqueKhaoula SettaraPas encore d'évaluation
- ANF RECIPROCS Juin2012 Affinement Poudres N Audebrand PDFDocument92 pagesANF RECIPROCS Juin2012 Affinement Poudres N Audebrand PDFDreamm AllPas encore d'évaluation
- Chapitre 1 DRXDocument45 pagesChapitre 1 DRXAyoub Djouabi100% (3)
- Defis Du CEA Infographie Cristallographie PDFDocument1 pageDefis Du CEA Infographie Cristallographie PDFAbderrahmane ChaterPas encore d'évaluation
- Chapitre I: Diffraction Des Rayons X Par La PoudreDocument15 pagesChapitre I: Diffraction Des Rayons X Par La PoudreBen othman AmalPas encore d'évaluation
- Cours Techniques DanalyseDocument116 pagesCours Techniques DanalyseNihad HaPas encore d'évaluation
- p1080 Caractérisation de Solides Cristallisés Par Diffraction XDocument18 pagesp1080 Caractérisation de Solides Cristallisés Par Diffraction XsalimPas encore d'évaluation
- Chap 12 ADocument13 pagesChap 12 ANorel HoudaPas encore d'évaluation
- Canne de RéfrigérationDocument3 pagesCanne de RéfrigérationAmjed LaritPas encore d'évaluation
- Cours Radiocristallographie s5 Pr-Tairi-Ppt (Mode de Compatibilité)Document189 pagesCours Radiocristallographie s5 Pr-Tairi-Ppt (Mode de Compatibilité)Lion LionPas encore d'évaluation
- Affinement de Profil Des Raies Par FullProf en Mode Profile Matching D'une HydroxyapatiteDocument37 pagesAffinement de Profil Des Raies Par FullProf en Mode Profile Matching D'une HydroxyapatiteBrahim Chafik El IdrissiPas encore d'évaluation
- Comportement Thermique Des Melanges Kaolin DolomieDocument8 pagesComportement Thermique Des Melanges Kaolin Dolomiekha elamPas encore d'évaluation
- PoudresDocument78 pagesPoudresMariam Rosa100% (1)
- 03 - Structure Cristalline Des SolidesDocument53 pages03 - Structure Cristalline Des Solidesstoph1290100% (1)
- Diffractionx PatrimoineDocument12 pagesDiffractionx PatrimoineFaissal El KhazantiPas encore d'évaluation
- Compléments de Cristallographie - F.hatert - 2013-2014Document47 pagesCompléments de Cristallographie - F.hatert - 2013-2014guijacko100% (1)