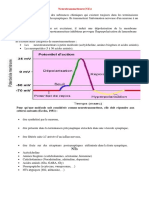
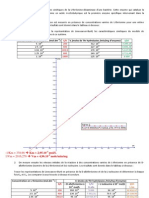
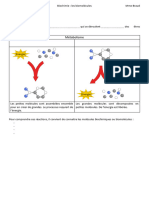
Vous aimerez peut-être aussi
- TD.02. BiomolDocument35 pagesTD.02. BiomolChawki Mokadem100% (1)
- TD N - 01 - Extraction Des Acides NucléiquesDocument21 pagesTD N - 01 - Extraction Des Acides NucléiquesHikari Kazue0% (2)
- Cours BM Master Les Techniques1Document29 pagesCours BM Master Les Techniques1Zineb IfriPas encore d'évaluation
- 1388 Outils de La Biologie Moleculaire M AberkaneDocument86 pages1388 Outils de La Biologie Moleculaire M AberkaneZakariya nadirPas encore d'évaluation
- Cour 1 EXTRACTION DES ANDocument48 pagesCour 1 EXTRACTION DES ANhissein saleh hisseinPas encore d'évaluation
- Polycopié de Travaux Pratiques Biologie Moléculaire: Universite Mohammed V - Agdal Faculte Des Sciences - RabatDocument11 pagesPolycopié de Travaux Pratiques Biologie Moléculaire: Universite Mohammed V - Agdal Faculte Des Sciences - RabatELGTAIBI HAJARPas encore d'évaluation
- Biologie Moleculaire - Chap1-Structure Des Acides NucleiquesDocument8 pagesBiologie Moleculaire - Chap1-Structure Des Acides NucleiquesMustapha SamiPas encore d'évaluation
- Hybridation Moléculaire - Acides NucléiquesDocument36 pagesHybridation Moléculaire - Acides NucléiquesMaï SsaPas encore d'évaluation
- Cours 04 L'hybridation MoléculaireDocument40 pagesCours 04 L'hybridation Moléculairegaming passionPas encore d'évaluation
- Génétique Microbienne PDFDocument65 pagesGénétique Microbienne PDFAnonymous MKSfyYyODPPas encore d'évaluation
- Diagnostic VirologiqueDocument5 pagesDiagnostic VirologiqueLylia Bessire100% (2)
- 7 - PCRDocument23 pages7 - PCRVino DongaPas encore d'évaluation
- Le Séquencage DADN 1Document4 pagesLe Séquencage DADN 1Kada BouameurPas encore d'évaluation
- 2 Canevas Licence - Biotechnologie MicrobienneDocument83 pages2 Canevas Licence - Biotechnologie MicrobienneHiba BourkabPas encore d'évaluation
- Culture CellulaireDocument25 pagesCulture CellulaireHadjer AdaidaPas encore d'évaluation
- TD Biologie Moléculaire BPR 2021Document21 pagesTD Biologie Moléculaire BPR 2021rafaramalala1307Pas encore d'évaluation
- Culture Cellulaire Et Virologie. Intérêts Et Applications de La Culture Cellulaire en VirologieDocument8 pagesCulture Cellulaire Et Virologie. Intérêts Et Applications de La Culture Cellulaire en Virologiemon ipadPas encore d'évaluation
- PCRDocument14 pagesPCRopenlabunistraPas encore d'évaluation
- Partie 1 - Biologie MoleculaireDocument48 pagesPartie 1 - Biologie MoleculaireZineb ZinebPas encore d'évaluation
- Genetique s5 TD 2Document15 pagesGenetique s5 TD 2dark kristalPas encore d'évaluation
- Exploration Du Metabolisme GlucidiqueDocument40 pagesExploration Du Metabolisme GlucidiqueAbdoulaye sebyPas encore d'évaluation
- TD Génomique Master UFR Sci Bio DR DagoDocument18 pagesTD Génomique Master UFR Sci Bio DR DagoPrince Otti's Officiel100% (1)
- Nutrition Croissance PharmacieDocument10 pagesNutrition Croissance PharmacieGarry AmneyPas encore d'évaluation
- ParasitologieDocument64 pagesParasitologieBouazzi IslemmPas encore d'évaluation
- TP - 01Document2 pagesTP - 01Slimkhane DzPas encore d'évaluation
- SCH Mas Cours SignalisationDocument35 pagesSCH Mas Cours SignalisationWael HahaPas encore d'évaluation
- Les Différents Types D'hybridation MoléculaireDocument10 pagesLes Différents Types D'hybridation Moléculairesherry1808Pas encore d'évaluation
- Parasitologie Générale 2023Document18 pagesParasitologie Générale 2023Flora NdouyangPas encore d'évaluation
- Genetique1an HybridationDocument26 pagesGenetique1an HybridationHaniDjekrifPas encore d'évaluation
- Biologie - Cours: 2: La Synthèse Des ProtéinesDocument37 pagesBiologie - Cours: 2: La Synthèse Des ProtéinesPharma1f2012100% (3)
- Chapitre V Virologie PDFDocument17 pagesChapitre V Virologie PDFbouguessa norelhoudaPas encore d'évaluation
- Cours de Génétique Humaine 1Document73 pagesCours de Génétique Humaine 1Naima BouhmaidaPas encore d'évaluation
- La PCR PDFDocument27 pagesLa PCR PDFfaroukPas encore d'évaluation
- Infectiologie Cours 28 Les Virus OncogenesDocument18 pagesInfectiologie Cours 28 Les Virus OncogenesPellePas encore d'évaluation
- Lpro TD 1 Oct16 PDFDocument10 pagesLpro TD 1 Oct16 PDFAnonymous FYcmbifCtmPas encore d'évaluation
- Examen Croissance PlantesDocument8 pagesExamen Croissance PlantesSalma Salmaaa100% (1)
- Diapos Ed9 ProteomiqueDocument55 pagesDiapos Ed9 ProteomiqueEmna El Hammi100% (1)
- Cours 4 Clonage MoleculaireDocument67 pagesCours 4 Clonage MoleculaireChawki MokademPas encore d'évaluation
- 13 - Classification Des Bactéries D'intérêt Médical - Les Bacilles À Gram (-) Non ExigeantsDocument56 pages13 - Classification Des Bactéries D'intérêt Médical - Les Bacilles À Gram (-) Non Exigeantsbouchakour meryemPas encore d'évaluation
- Examen Bactériologique Des Selles (Coproculture) - LAPTOP-L6BKA9FMDocument45 pagesExamen Bactériologique Des Selles (Coproculture) - LAPTOP-L6BKA9FMSahbi Mehrzi100% (1)
- CR 8 - Cytogénétique MoléculaireDocument51 pagesCR 8 - Cytogénétique MoléculaireMouad HiliaPas encore d'évaluation
- BIO 304 - ELECTROPHORESE - Cours 2022Document30 pagesBIO 304 - ELECTROPHORESE - Cours 2022mistic JayPas encore d'évaluation
- Les Virus 3A MED .10.04..2022Document33 pagesLes Virus 3A MED .10.04..2022Ker MichePas encore d'évaluation
- Les Protozooses 2A Bio 2018Document31 pagesLes Protozooses 2A Bio 2018حمزة بن أحمدPas encore d'évaluation
- Cours 8 PCRDocument47 pagesCours 8 PCRHikari KazuePas encore d'évaluation
- Hybridation MoléculaireDocument2 pagesHybridation MoléculaireHÄ ÝÄŤ100% (1)
- TD3 BV T.conducteur IDocument6 pagesTD3 BV T.conducteur IRahma LionnePas encore d'évaluation
- Methode de Diagnostic 10Document55 pagesMethode de Diagnostic 10MACON824100% (1)
- Elisa 2Document5 pagesElisa 2rawaabdallahPas encore d'évaluation
- La Génétique en Poche - Dr. Loucif & Dr. LaaouadDocument21 pagesLa Génétique en Poche - Dr. Loucif & Dr. LaaouadWassila HallouPas encore d'évaluation
- TD Génomique Master UFR Sci Bio DR DagoDocument11 pagesTD Génomique Master UFR Sci Bio DR DagoFANKELE ALASSANE KONE100% (1)
- TP4 Microbiologie Générale-Identification Par Caractérisation BiochimiqueDocument11 pagesTP4 Microbiologie Générale-Identification Par Caractérisation Biochimiquesonia HALAOULIPas encore d'évaluation
- !!!!!!!!cours Biologie Moleculaie S5Document46 pages!!!!!!!!cours Biologie Moleculaie S5Miro100% (1)
- 17 - Mycobacterium TuberculosisDocument33 pages17 - Mycobacterium Tuberculosissaydali dzPas encore d'évaluation
- Hémoglobine Méthode Colorimétrique (Cyanméthémoglobine) 1Document2 pagesHémoglobine Méthode Colorimétrique (Cyanméthémoglobine) 1zlimitoune0% (2)
- Southern BlotDocument5 pagesSouthern BlotNina AbPas encore d'évaluation
- CHAPITRE 2 BIOLOGIE MOLECULAIRE Mme OUNISDocument18 pagesCHAPITRE 2 BIOLOGIE MOLECULAIRE Mme OUNISZineb ZinebPas encore d'évaluation
- Cour de Virologie PDFDocument21 pagesCour de Virologie PDFysanPas encore d'évaluation
- Genie Genetique2020Document17 pagesGenie Genetique2020Abdoul karim SamakePas encore d'évaluation
- Métabolisme de BaseDocument6 pagesMétabolisme de BaseaxelPas encore d'évaluation
- FDocument3 pagesFI'm DekuuPas encore d'évaluation
- 1 Proteines 2007-08Document23 pages1 Proteines 2007-08anon_846940093Pas encore d'évaluation
- BIOENERGETIQUEDocument71 pagesBIOENERGETIQUEkhalilchips2005Pas encore d'évaluation
- 1-TD Groupe 2Document4 pages1-TD Groupe 2MariettePas encore d'évaluation
- EXERCICE 01 - CopieDocument5 pagesEXERCICE 01 - CopieIman BelhadiaPas encore d'évaluation
- Chapitre II Expression Des Gènes (Transcription Et Traduction) Leur RégulationDocument9 pagesChapitre II Expression Des Gènes (Transcription Et Traduction) Leur Régulationsolo gamer50% (2)
- C11. Physiologie Du Pancéas EndocrineDocument8 pagesC11. Physiologie Du Pancéas EndocrineAssarm Albattar KikoPas encore d'évaluation
- Chapitre 5-La Glycolyse (Voie D'embden-Meyeroff-Parnas)Document17 pagesChapitre 5-La Glycolyse (Voie D'embden-Meyeroff-Parnas)stephen njemsPas encore d'évaluation
- 2.oses - MorelDocument32 pages2.oses - Morelsoumia lefkirPas encore d'évaluation
- 5 AntigèneDocument5 pages5 AntigèneOsama O-bPas encore d'évaluation
- Les Etapes de La GlycolyseDocument7 pagesLes Etapes de La GlycolyseNaruto & SasukePas encore d'évaluation
- Communication CellulaireDocument22 pagesCommunication CellulaireAmïn Fïlalï100% (2)
- Bord G2Document152 pagesBord G2AngelaPas encore d'évaluation
- Recherche DES HemolysinesDocument3 pagesRecherche DES HemolysinesAhmed MokhtariPas encore d'évaluation
- 2) Exercices Structure Et Organisation de l'ADNDocument17 pages2) Exercices Structure Et Organisation de l'ADNOne Love LifePas encore d'évaluation
- Cours Chapitre 2 Membrane PlasmiqueDocument5 pagesCours Chapitre 2 Membrane Plasmiqueayman HammoudaPas encore d'évaluation
- Series TD GenetiqueDocument36 pagesSeries TD GenetiqueNouhoum TraoréPas encore d'évaluation
- ÉlectroDocument13 pagesÉlectroOTOTEPas encore d'évaluation
- Neuro 2Document12 pagesNeuro 2tinaPas encore d'évaluation
- Chapitre 11 - L'appareil EndocrinienDocument30 pagesChapitre 11 - L'appareil EndocrinienBaptiste LetorPas encore d'évaluation
- Métabolisme LipidesDocument51 pagesMétabolisme Lipidesscaren efona100% (1)
- S1 TD2 Protides PDFDocument8 pagesS1 TD2 Protides PDFmartinmune67% (3)
- TSTL - Bioch.Cours .Chap.2.Enzymologie - Correction Exercice.7Document3 pagesTSTL - Bioch.Cours .Chap.2.Enzymologie - Correction Exercice.7Chris Zericho0% (3)
- (EMC - Biologie Médicale 2006-Jan Vol. 1 Iss. 1) Myara, Jacques - Cholestérol Total (2006) (10.1016 - s2211-9698 (06) 76045-9) - Libgen - LiDocument4 pages(EMC - Biologie Médicale 2006-Jan Vol. 1 Iss. 1) Myara, Jacques - Cholestérol Total (2006) (10.1016 - s2211-9698 (06) 76045-9) - Libgen - LiNéfer TitiPas encore d'évaluation
- Le Noyau SciencesDocument2 pagesLe Noyau SciencesGauthier MehaudensPas encore d'évaluation
- Introduction + Aa - ElèveDocument8 pagesIntroduction + Aa - Elèvesoufireda25Pas encore d'évaluation
- Genie Genetique Chapitre 3Document5 pagesGenie Genetique Chapitre 3Ñoor ĒllîzzaPas encore d'évaluation
- Vitamines B TableauDocument2 pagesVitamines B TableauMarouaneBouhabsPas encore d'évaluation
- Université Ibn Tofai1Document3 pagesUniversité Ibn Tofai1AMi NEPas encore d'évaluation