Vous aimerez peut-être aussi
- Corrigé Centrale 2010 Acide OxaliqueDocument6 pagesCorrigé Centrale 2010 Acide OxaliquePascal FrajmanPas encore d'évaluation
- Exercices-Materiaux-Chapitre IDocument8 pagesExercices-Materiaux-Chapitre IJewel Slam100% (1)
- SMDocument26 pagesSMLghazi OumssadPas encore d'évaluation
- 2010 Pondichery Exo1 Correction Polonium 6ptsDocument2 pages2010 Pondichery Exo1 Correction Polonium 6ptsla physique selon le programme FrançaisPas encore d'évaluation
- 2013 Antilles Exo2 Correction Pommes 10ptsDocument3 pages2013 Antilles Exo2 Correction Pommes 10ptsYasmine HamichePas encore d'évaluation
- Corrigé Serie D Session 2015Document8 pagesCorrigé Serie D Session 2015inayalinah4Pas encore d'évaluation
- DS4 - Reactions Nucleaires - Cohésion Solides - DissolutionDocument3 pagesDS4 - Reactions Nucleaires - Cohésion Solides - DissolutionKARENPas encore d'évaluation
- Liaison ChimiqueDocument31 pagesLiaison Chimiquezdramane950Pas encore d'évaluation
- Controle 2020 QuantiqueDocument2 pagesControle 2020 QuantiqueRhm Gaming100% (1)
- Exercices Corriges Equilibrer Des Reactions Nucleaires Reaction de Fusion Dans Le SoleilDocument3 pagesExercices Corriges Equilibrer Des Reactions Nucleaires Reaction de Fusion Dans Le Soleilroudy100% (1)
- ch01 SolutionnaireDocument40 pagesch01 SolutionnaireWaqar BaigPas encore d'évaluation
- 5 - Effets ÉlectriquesDocument12 pages5 - Effets Électriqueshoussam.mouhtadiPas encore d'évaluation
- Atomistique Ex CorDocument2 pagesAtomistique Ex Coramirtrk80100% (1)
- Physique 2011 Principale Correction PDFDocument9 pagesPhysique 2011 Principale Correction PDFRoua ManiPas encore d'évaluation
- Alcènes Énoncés ExercicesDocument62 pagesAlcènes Énoncés ExercicesMaxime EidelsbergPas encore d'évaluation
- Cours Orga SMC S3 2021Document118 pagesCours Orga SMC S3 2021zniber.fatima2001Pas encore d'évaluation
- Corrigé Serie D 2021Document7 pagesCorrigé Serie D 2021ChristellePas encore d'évaluation
- PC 1D 1a Ta S2 2024Document4 pagesPC 1D 1a Ta S2 2024arsene BATAWUILAPas encore d'évaluation
- Examen National Physique Chimie SPC 2016 Normale Corrige 1Document9 pagesExamen National Physique Chimie SPC 2016 Normale Corrige 1othmane GbPas encore d'évaluation
- Devoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2013-2014)Document5 pagesDevoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2013-2014)AliMchirguiPas encore d'évaluation
- Corrigé CNC Chimie 2012 MPDocument6 pagesCorrigé CNC Chimie 2012 MPzazazaz2000100% (1)
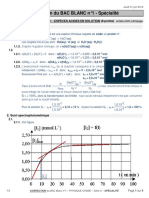
- TS - BAC Blanc N°1 Corrigé - Spécialité PDFDocument4 pagesTS - BAC Blanc N°1 Corrigé - Spécialité PDFphytanjaPas encore d'évaluation
- Corrigé de L'épreuve de Sciences Physiques: Universite C A D DDocument5 pagesCorrigé de L'épreuve de Sciences Physiques: Universite C A D DLindeltaylor DioufPas encore d'évaluation
- Physique C PDFDocument4 pagesPhysique C PDFالغزيزال الحسن EL GHZIZAL HassanePas encore d'évaluation
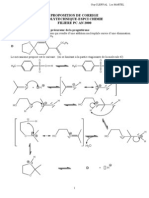
- Architecture Moléculaire Et StéréochimieDocument5 pagesArchitecture Moléculaire Et StéréochimielucatelliPas encore d'évaluation
- Concours 2012 (+sol.) Proposition EPST23Document19 pagesConcours 2012 (+sol.) Proposition EPST23Nidal BestPas encore d'évaluation
- ExamenDocument3 pagesExamenIheb DassiPas encore d'évaluation
- 1es Chap 1 Cours Poly CompleteDocument6 pages1es Chap 1 Cours Poly CompleteDavid LandellePas encore d'évaluation
- C 00 XP 1 CDocument13 pagesC 00 XP 1 CMihnea GamanPas encore d'évaluation
- 5351 Entrainement TSDocument6 pages5351 Entrainement TSmohammed laadili100% (1)
- 2010 Asie Exo3 Correction QROCChimie 6 5pts - 2Document3 pages2010 Asie Exo3 Correction QROCChimie 6 5pts - 2Youssef DahaniPas encore d'évaluation
- Corrige td1 ElectrochimieDocument7 pagesCorrige td1 Electrochimieayoub dahbi100% (1)
- CNC 2000 Chimie MP CorrectionDocument4 pagesCNC 2000 Chimie MP CorrectionNouhaila AfakirePas encore d'évaluation
- Sujets Pass2021Document4 pagesSujets Pass2021indien cheperPas encore d'évaluation
- Devoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2014-2015) MR Handoura NaceurDocument4 pagesDevoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2014-2015) MR Handoura NaceurMohamed SaidiPas encore d'évaluation
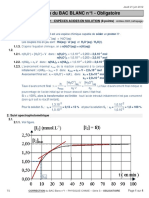
- TS - BAC Blanc N°1 Corrigé - ObligatoireDocument4 pagesTS - BAC Blanc N°1 Corrigé - ObligatoirephytanjaPas encore d'évaluation
- Devoir Up PhysiqueDocument4 pagesDevoir Up PhysiquedianechibidouxPas encore d'évaluation
- Bacc PC Serie C 1999 - 2013Document55 pagesBacc PC Serie C 1999 - 2013riantsoaPas encore d'évaluation
- 1cm41mch1 245884Document5 pages1cm41mch1 245884Absa NdiayePas encore d'évaluation
- C Chtsi2021Document8 pagesC Chtsi2021Rose kalvine MougoulaPas encore d'évaluation
- Examen1 Réactivité Chimique FST-MeknèsDocument6 pagesExamen1 Réactivité Chimique FST-MeknèsayabourhdifaPas encore d'évaluation
- Hückel Elèves TDDocument3 pagesHückel Elèves TDami rPas encore d'évaluation
- Correction TD Chimie Générale Fiche1Document6 pagesCorrection TD Chimie Générale Fiche1comboudriPas encore d'évaluation
- Chap 4 EFFET S ELECTRONIQUESDocument8 pagesChap 4 EFFET S ELECTRONIQUESEL Menani NadiaPas encore d'évaluation
- Bac PCDocument5 pagesBac PCOumar KakoroPas encore d'évaluation
- Chimie-Bioch 1 2010Document17 pagesChimie-Bioch 1 2010raspberry najmPas encore d'évaluation
- Edlab PC 2017 CorreDocument2 pagesEdlab PC 2017 Corresaidouly95Pas encore d'évaluation
- Corrige Bac 2009Document2 pagesCorrige Bac 2009Claudy TsakengPas encore d'évaluation
- Devoir Corrigé de Synthèse N°3 - Physique - Bac Mathématiques (2010-2011) Elève SindaDocument7 pagesDevoir Corrigé de Synthèse N°3 - Physique - Bac Mathématiques (2010-2011) Elève SindaTawfiq Weld EL ArbiPas encore d'évaluation
- Chapitre IVDocument8 pagesChapitre IVYu-oh CrysisPas encore d'évaluation
- Cu Ag MG Cu Ag MG POCDocument3 pagesCu Ag MG Cu Ag MG POCorsiny clinton TCHAPTCHETPas encore d'évaluation
- TD Avec SolutionDocument21 pagesTD Avec SolutionAll AhmeDcia0% (1)
- Mecanisme Reactionnel SN1 SN2 OuziDocument33 pagesMecanisme Reactionnel SN1 SN2 Ouzisafae ziyatiPas encore d'évaluation
- DS05Document11 pagesDS05baatoutmbPas encore d'évaluation
- Effets Electroniques OrganiqueDocument21 pagesEffets Electroniques OrganiqueLahcen ElamraouiPas encore d'évaluation
- Homework Corrosion BekhdidjaDocument4 pagesHomework Corrosion BekhdidjaفتحيPas encore d'évaluation
- Cor Sujet ChimieDocument5 pagesCor Sujet ChimiefaridattallahPas encore d'évaluation
- Physique Bac MathDocument4 pagesPhysique Bac MathSofia BahriniPas encore d'évaluation
- Cours Techniques SpectroscopiquesDocument34 pagesCours Techniques Spectroscopiquesami rPas encore d'évaluation
- Examen Chimie Theorique SMC5 - Partie HuckelDocument4 pagesExamen Chimie Theorique SMC5 - Partie Huckelami rPas encore d'évaluation
- 3.systemes Polyelectroniques AtomesDocument21 pages3.systemes Polyelectroniques Atomesami rPas encore d'évaluation
- APCQ3Document16 pagesAPCQ3ami rPas encore d'évaluation
- Cours CQ CIDocument86 pagesCours CQ CIami rPas encore d'évaluation
- Simple Huckel CH404Document3 pagesSimple Huckel CH404ami rPas encore d'évaluation
- Chap3 Modele Ondulatoire de L'atomeDocument15 pagesChap3 Modele Ondulatoire de L'atomeami rPas encore d'évaluation
- 2021 sp1 Devoir01Document3 pages2021 sp1 Devoir01ami rPas encore d'évaluation
- Décoloration Du PH - PHDocument2 pagesDécoloration Du PH - PHami rPas encore d'évaluation
- 2014 01 26 TD3Document2 pages2014 01 26 TD3ami rPas encore d'évaluation
- امن المخبرDocument1 pageامن المخبرami rPas encore d'évaluation
- Hückel Elèves TDDocument3 pagesHückel Elèves TDami rPas encore d'évaluation
- Travaux Pratiques de Thermodynamique Et de Cinétique ChimiquesDocument18 pagesTravaux Pratiques de Thermodynamique Et de Cinétique Chimiquesami rPas encore d'évaluation
- Expériences ChimieDocument9 pagesExpériences Chimieami rPas encore d'évaluation
- Présentation Teneur en Eau Dans Les Huiles DiélctriquesDocument5 pagesPrésentation Teneur en Eau Dans Les Huiles Diélctriqueschichid2008Pas encore d'évaluation
- Extraction Liquide Liquide 20 - 210517 - 114623Document24 pagesExtraction Liquide Liquide 20 - 210517 - 114623Mahmoud MazboudiPas encore d'évaluation
- Extraction Liquide-LiquideDocument7 pagesExtraction Liquide-LiquideAbdou MiringhiPas encore d'évaluation
- UE CHM 4168 2022-2023 TD No1 SyntheseDocument2 pagesUE CHM 4168 2022-2023 TD No1 SyntheseIsocrate DourrawaPas encore d'évaluation
- TS - BAC Blanc N°1 Corrigé - Spécialité PDFDocument4 pagesTS - BAC Blanc N°1 Corrigé - Spécialité PDFphytanjaPas encore d'évaluation
- DPS Iii - Q - Cis - Apd - CVD - Clim-Fte - Syn - Bat.02Document9 pagesDPS Iii - Q - Cis - Apd - CVD - Clim-Fte - Syn - Bat.02valdes zebazePas encore d'évaluation
- .Parties Du DiscoursDocument8 pages.Parties Du DiscoursKirby KjongPas encore d'évaluation
- Chapitre III Transitions Du Premier Et Second OrdreDocument13 pagesChapitre III Transitions Du Premier Et Second OrdreLamiae NouiguiPas encore d'évaluation
- Le Théâtre Classique, Module 18, Semestre 3, Aicha BouraisDocument7 pagesLe Théâtre Classique, Module 18, Semestre 3, Aicha BouraisAdil AmmariPas encore d'évaluation
- Des Melanges DangereuxDocument4 pagesDes Melanges Dangereuxproduction.assur2cPas encore d'évaluation
- Exercices 13 Suivi TemporelDocument1 pageExercices 13 Suivi Temporelالغزيزال الحسن EL GHZIZAL Hassane75% (4)
- Théorie Du Champ Cristallin 2019-2020Document2 pagesThéorie Du Champ Cristallin 2019-2020Roi AroufPas encore d'évaluation
- Fiche 1 - Principales Propriétés Des Différentes Classes de PolymèresDocument26 pagesFiche 1 - Principales Propriétés Des Différentes Classes de PolymèresManamba Dite Sisse TraoréPas encore d'évaluation
- DS2 3 Eme T 2023Document4 pagesDS2 3 Eme T 2023Riadh MarouaniPas encore d'évaluation
- Indicateurs ColoresDocument3 pagesIndicateurs ColoresWEMBE RAOULPas encore d'évaluation
- Xypex ConcentreDocument4 pagesXypex ConcentreAdel BouatayPas encore d'évaluation
- Sujet2015 Cap CDocument10 pagesSujet2015 Cap CpaulardPas encore d'évaluation
- Activité 3: L'élément Oxygène Dans Notre Atmosphère (Correction)Document2 pagesActivité 3: L'élément Oxygène Dans Notre Atmosphère (Correction)EvaPas encore d'évaluation
- Exploration Des Ressources Minerales Et Energetiques Bouabsa LDocument79 pagesExploration Des Ressources Minerales Et Energetiques Bouabsa LMohammed Larbi FaradjiPas encore d'évaluation
- TP Comment Synthétiser Et Caractériser Un PolymèreDocument3 pagesTP Comment Synthétiser Et Caractériser Un PolymèrealexbardonfrPas encore d'évaluation
- Projet DeclairageDocument7 pagesProjet Declairageabdelbasset elwaddaniPas encore d'évaluation
- Les Matériaux Nanostructurés PDFDocument49 pagesLes Matériaux Nanostructurés PDFmortadaPas encore d'évaluation
- 5-Structures AlternativesDocument16 pages5-Structures AlternativesOthman ShonePas encore d'évaluation
- Travaux Dirigés de Chimie N° 6: Exercice 1: Équations de Réaction Et Constantes D'équilibreDocument4 pagesTravaux Dirigés de Chimie N° 6: Exercice 1: Équations de Réaction Et Constantes D'équilibreAbderrahman IGHNIHPas encore d'évaluation
- Correction Exercices Chapitre N°8 - Oxydo-Réduction Dans Les Piles Et AccumulateursDocument3 pagesCorrection Exercices Chapitre N°8 - Oxydo-Réduction Dans Les Piles Et Accumulateursunsheatedsword55Pas encore d'évaluation
- Salim DouaaDocument220 pagesSalim DouaaAbdoPas encore d'évaluation
- InmunohistochimieDocument4 pagesInmunohistochimiepame RodriguezPas encore d'évaluation
- Evaluation Solides Cristallins Sujet A À FDocument10 pagesEvaluation Solides Cristallins Sujet A À Frania rejebPas encore d'évaluation
- Mig225syn FRDocument18 pagesMig225syn FRalain.broussyPas encore d'évaluation