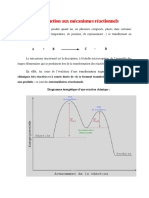
Vous aimerez peut-être aussi
- Effets Inducteurs Ou Inductifs Et Mesomeres Cours1Document15 pagesEffets Inducteurs Ou Inductifs Et Mesomeres Cours1zahira rajebPas encore d'évaluation
- Effets Electroniques PDFDocument14 pagesEffets Electroniques PDFmohsatPas encore d'évaluation
- Effets Electroniques PDFDocument4 pagesEffets Electroniques PDFAllalPas encore d'évaluation
- ChimieDocument9 pagesChimiebchaimaa747Pas encore d'évaluation
- Effets ÉlectroniquesDocument13 pagesEffets ÉlectroniquesTarik ZaidPas encore d'évaluation
- Effets Électroniques Et Mécanismes RéactionnelsDocument11 pagesEffets Électroniques Et Mécanismes Réactionnelsdaniel.bignoumbaPas encore d'évaluation
- Chap IVDocument32 pagesChap IVMamadou lamine DiattaPas encore d'évaluation
- Chapitre Iii Et Chapitre Iv Cours de Esb 132 2022-2023Document38 pagesChapitre Iii Et Chapitre Iv Cours de Esb 132 2022-2023laminoumhd75Pas encore d'évaluation
- Chapitre IV-2020-Les Effets ÉlectroniquesDocument41 pagesChapitre IV-2020-Les Effets ÉlectroniquesSoufiane RouchadPas encore d'évaluation
- Chap 4 EFFET S ELECTRONIQUESDocument8 pagesChap 4 EFFET S ELECTRONIQUESEL Menani NadiaPas encore d'évaluation
- REVISION Chimie Orga Ecole (Enregistrement Automatique)Document28 pagesREVISION Chimie Orga Ecole (Enregistrement Automatique)Charaf SekhsoukhPas encore d'évaluation
- Chap 3Document46 pagesChap 3emmanuel kenfackPas encore d'évaluation
- 5 - Effets ÉlectriquesDocument12 pages5 - Effets Électriqueshoussam.mouhtadiPas encore d'évaluation
- Effets Electroniques OrganiqueDocument21 pagesEffets Electroniques OrganiqueLahcen ElamraouiPas encore d'évaluation
- Les Effets ElectroniquesDocument8 pagesLes Effets ElectroniquesChTri Lyrics100% (1)
- Chapitre 1Document42 pagesChapitre 1Mohamed mePas encore d'évaluation
- Chapitre IV - E - ElectroniqueDocument15 pagesChapitre IV - E - Electroniquetatatadadada47Pas encore d'évaluation
- Chapitre Vii - Effets Electroniques-2Document15 pagesChapitre Vii - Effets Electroniques-2adnan aitlahcPas encore d'évaluation
- Cours - Liaison - Chim ChapI - II - ElalemDocument18 pagesCours - Liaison - Chim ChapI - II - ElalemFatima EL AROUSSIPas encore d'évaluation
- Chapitre Effets Électroniques 2Document16 pagesChapitre Effets Électroniques 2Ismail ZitouniPas encore d'évaluation
- Effets Electroniques Et Introduction Aux Mécanisme RéactionnelsDocument28 pagesEffets Electroniques Et Introduction Aux Mécanisme RéactionnelsbouaddouanisPas encore d'évaluation
- Chapitre IVDocument8 pagesChapitre IVYu-oh CrysisPas encore d'évaluation
- Effets Inductifs Et MesomeresDocument5 pagesEffets Inductifs Et MesomeresJunior ALLODJIPas encore d'évaluation
- La Liaison Chimique 2020 2021Document16 pagesLa Liaison Chimique 2020 2021ayachelayanePas encore d'évaluation
- Effets - Lectroniques - Chimie - Organique - Mon Cours2Document4 pagesEffets - Lectroniques - Chimie - Organique - Mon Cours2meyemarion99Pas encore d'évaluation
- Effet Electronique 22Document76 pagesEffet Electronique 22squalli.hachemPas encore d'évaluation
- Cours Pharmacie Les Effets ÉlectroniquesDocument18 pagesCours Pharmacie Les Effets ÉlectroniquesMira AmiraPas encore d'évaluation
- Cours COII - SN PDFDocument65 pagesCours COII - SN PDFYoussef FihriPas encore d'évaluation
- CHAPITRE I Spectrophotométrie UV VisibleDocument12 pagesCHAPITRE I Spectrophotométrie UV VisibleMouka MoukaPas encore d'évaluation
- 06-La Liaison Chimique 2020-2021Document13 pages06-La Liaison Chimique 2020-2021Walid AzzamPas encore d'évaluation
- Devoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2013-2014)Document5 pagesDevoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2013-2014)AliMchirguiPas encore d'évaluation
- Travaux Dirigés - Série N°4Document27 pagesTravaux Dirigés - Série N°4Labrim YassinePas encore d'évaluation
- Difo - CHAPITREIV - EFFET ELECTRDocument24 pagesDifo - CHAPITREIV - EFFET ELECTRDifo Voukang HarounaPas encore d'évaluation
- Chap 5Document7 pagesChap 5ali akredPas encore d'évaluation
- Laisson Chimiques SMPC S2Document37 pagesLaisson Chimiques SMPC S2Saidi AbdelghaniPas encore d'évaluation
- Chapitre Iv-Chimie 4 PDFDocument5 pagesChapitre Iv-Chimie 4 PDFFerielPas encore d'évaluation
- Serie 1 2022 23Document8 pagesSerie 1 2022 23mariem OuriPas encore d'évaluation
- Chimie Des Electrolytes CompressedDocument31 pagesChimie Des Electrolytes CompressedDjemil HalimaPas encore d'évaluation
- Effets ElectroniquesDocument7 pagesEffets ElectroniquesAbdoul karim SamakePas encore d'évaluation
- Correction TD Chimie Générale Fiche1Document6 pagesCorrection TD Chimie Générale Fiche1comboudriPas encore d'évaluation
- ElectrochimieDocument32 pagesElectrochimiehadil.23sd56Pas encore d'évaluation
- Chapitre IVDocument11 pagesChapitre IVbousnane3bousnanePas encore d'évaluation
- Chimie 1. Bases de La ChimieDocument50 pagesChimie 1. Bases de La Chimieexaucemakizodila60Pas encore d'évaluation
- Chimie-Bioch 1 2010Document17 pagesChimie-Bioch 1 2010raspberry najmPas encore d'évaluation
- TD de Chimie GeneraleDocument3 pagesTD de Chimie GeneralewilliammerlindonfackPas encore d'évaluation
- Effets de StructureDocument10 pagesEffets de Structurezri zrPas encore d'évaluation
- 3 - Chap3 - Réactions Acide-Base - Mode de Compatibilité PDFDocument53 pages3 - Chap3 - Réactions Acide-Base - Mode de Compatibilité PDFoulai100% (1)
- Cours de Chimie OrganiqueDocument28 pagesCours de Chimie Organiquedamn tweetsPas encore d'évaluation
- Électrochimie Chap-1Document17 pagesÉlectrochimie Chap-1Zineb SassiPas encore d'évaluation
- Cours Chimie Organique 2 (S4) TC BCG-convertiDocument126 pagesCours Chimie Organique 2 (S4) TC BCG-convertiRim ElmoutaoukkilPas encore d'évaluation
- Liaisons ChimiquesDocument28 pagesLiaisons ChimiquesMamineTecDelmaPas encore d'évaluation
- Cours Chimie OrganiqueDocument60 pagesCours Chimie OrganiqueFarah BahriPas encore d'évaluation
- 1 Cours Physico Chimie Des Électrolytes 2013Document31 pages1 Cours Physico Chimie Des Électrolytes 2013aouPas encore d'évaluation
- TD3 Structure - 12 - 13Document7 pagesTD3 Structure - 12 - 13sidi mohamed el amine nekkal100% (1)
- l2 - Cours1 Oxydoreduction Fevrier 2019Document23 pagesl2 - Cours1 Oxydoreduction Fevrier 2019Alexandre Kpangny BéniPas encore d'évaluation
- Introduction à la physique de la matièreD'EverandIntroduction à la physique de la matièreÉvaluation : 3 sur 5 étoiles3/5 (1)
- Cours Radiocristallographie Et Cristallochimie II SMC s5Document203 pagesCours Radiocristallographie Et Cristallochimie II SMC s5khadija limamPas encore d'évaluation
- Management Affirmation QuestionnaireDocument5 pagesManagement Affirmation QuestionnaireBenoit bbnPas encore d'évaluation
- Mécanisme de RéactionDocument8 pagesMécanisme de Réactionsafae ziyatiPas encore d'évaluation
- Cours La Cinétique FormelleDocument7 pagesCours La Cinétique Formellesafae ziyatiPas encore d'évaluation
- Mécanisme D'élimination E2Document8 pagesMécanisme D'élimination E2benjamin.arfi06Pas encore d'évaluation
- 6 Electrochimie Exercices Eleves CorrectionDocument13 pages6 Electrochimie Exercices Eleves CorrectionNikiemaPas encore d'évaluation
- Chimie Organique MécanismesDocument17 pagesChimie Organique MécanismesMLAN HesnaPas encore d'évaluation
- Reactions D EliminationDocument9 pagesReactions D Eliminationnini100% (1)
- Chimie Organique Fonctionnelle TD Corr 01Document2 pagesChimie Organique Fonctionnelle TD Corr 01123456789Pas encore d'évaluation
- Partiel Correction L1 Chimie Organique 2006 3Document6 pagesPartiel Correction L1 Chimie Organique 2006 3R-winPas encore d'évaluation
- Reaction Sub-ElimDocument37 pagesReaction Sub-ElimRima LettreuchPas encore d'évaluation
- 2 Addition Electrophile 2Document10 pages2 Addition Electrophile 2Bibi BibaPas encore d'évaluation
- Chapitre5 1Document9 pagesChapitre5 1Karim Radi100% (1)
- Corrigés Leçon 11 - Classification QuantitativeDocument4 pagesCorrigés Leçon 11 - Classification Quantitativeholyeric50Pas encore d'évaluation
- TD Intermediaires Reactionnels 2 SolutionDocument2 pagesTD Intermediaires Reactionnels 2 SolutionIssa SINDEPas encore d'évaluation
- Chapitre1 Chimie Organique 2Document8 pagesChapitre1 Chimie Organique 2jean luc abdias konanPas encore d'évaluation
- TD - Chimie Organique-Série 7 - Correction - Séance Du 09-05-23 PDFDocument19 pagesTD - Chimie Organique-Série 7 - Correction - Séance Du 09-05-23 PDFeya bmPas encore d'évaluation
- MécanismeDocument6 pagesMécanismeFarouk MessaoudPas encore d'évaluation
- Chim311-2021-Mécanismes RéactionelsDocument51 pagesChim311-2021-Mécanismes RéactionelsKornichonPas encore d'évaluation
- Boumendjel Ahcene p12 PDFDocument45 pagesBoumendjel Ahcene p12 PDFMàâçhë ĶămęłPas encore d'évaluation
- Chapitre 7 Chimie OrganiqueDocument46 pagesChapitre 7 Chimie OrganiqueleloPas encore d'évaluation
- Exercices Reaction ChimiqueDocument9 pagesExercices Reaction ChimiquePride Algerian ツPas encore d'évaluation
- Réactions de Substitution NucléophileDocument7 pagesRéactions de Substitution Nucléophilewarda MaPas encore d'évaluation
- Chap 3Document32 pagesChap 3Taha DaoudPas encore d'évaluation
- Halogenure AlkyleDocument21 pagesHalogenure AlkyleGrace BendaPas encore d'évaluation
- Chimie Organique Fonctionnelle TD Corr 08Document3 pagesChimie Organique Fonctionnelle TD Corr 08Kouadio Franck Tomasino KouamePas encore d'évaluation
- Chapitre 2 - Equilibrage Des Reactions RedoxDocument2 pagesChapitre 2 - Equilibrage Des Reactions RedoxJoe FinianosPas encore d'évaluation
- Chimie Organique Fonctionnelle TD Corr 05Document5 pagesChimie Organique Fonctionnelle TD Corr 05Khaoula TimliltPas encore d'évaluation
- Cours Les Dérivés Halogénés LCE2Document32 pagesCours Les Dérivés Halogénés LCE2Wissal ChammakhiPas encore d'évaluation
- Correction Examen 2014-2015Document4 pagesCorrection Examen 2014-2015api-497754935Pas encore d'évaluation
- Boumendjel Ahcene p06Document36 pagesBoumendjel Ahcene p06Chimiste ChimistePas encore d'évaluation
- TD Substitution Nucleophile Corrige 4Document6 pagesTD Substitution Nucleophile Corrige 4Daniel Nd100% (1)
- Chapitre V Partie 2Document18 pagesChapitre V Partie 2Missou PrincessePas encore d'évaluation
- Chapitre-Intermédiaires RéactionnelsDocument9 pagesChapitre-Intermédiaires RéactionnelsIsmail ZitouniPas encore d'évaluation