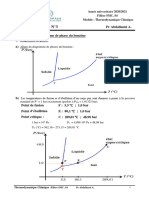
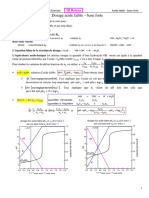
Vous aimerez peut-être aussi
- Corrigé - Serie - Slater - 2021Document11 pagesCorrigé - Serie - Slater - 2021Rawda Kh86% (7)
- Cours M2 Distil Complexe PDFDocument26 pagesCours M2 Distil Complexe PDFAyoub Magroud100% (2)
- Chapitre I (Potentiel Chimique) 2015Document37 pagesChapitre I (Potentiel Chimique) 2015Chaouki100% (1)
- Mélange VapeurDocument12 pagesMélange VapeurRawda KhPas encore d'évaluation
- Cours DistillDocument25 pagesCours DistillalexisbradPas encore d'évaluation
- Cours PetrogeochimieDocument48 pagesCours Petrogeochimiehamza manafPas encore d'évaluation
- TD22Document2 pagesTD22Saliou SENEPas encore d'évaluation
- L3 DistillationDocument80 pagesL3 DistillationLeila Ben HassinePas encore d'évaluation
- Doc1 Mlanges Binaires - Liq SolDocument7 pagesDoc1 Mlanges Binaires - Liq Solt123medPas encore d'évaluation
- Compressibilité de L'eauDocument1 pageCompressibilité de L'eauzohir AlouachePas encore d'évaluation
- Rattrapage S4 - Octobre 2020-SolutionDocument4 pagesRattrapage S4 - Octobre 2020-Solutionyehya boureghdaPas encore d'évaluation
- Resume Cours FluideDocument5 pagesResume Cours FluideSerigne Saliou NgomPas encore d'évaluation
- D4.13.Ch4.machine Frigorifique bts02 CorrigeDocument4 pagesD4.13.Ch4.machine Frigorifique bts02 CorrigewafaPas encore d'évaluation
- Mecanique Des Fluides Cours 05 0030 0030Document1 pageMecanique Des Fluides Cours 05 0030 0030Funny ChildrenPas encore d'évaluation
- Les Diagrammes ThermodynamiquesDocument3 pagesLes Diagrammes ThermodynamiquesHassan Amrani0% (1)
- DistillationDocument16 pagesDistillationAhmad TlilaniPas encore d'évaluation
- Rappel - ThermoDocument18 pagesRappel - ThermoEmmanuel smith TaïgaPas encore d'évaluation
- Mecanique Des Fluide Master24Document136 pagesMecanique Des Fluide Master24aminaidrissi804Pas encore d'évaluation
- Série #3 - S4 2021 - SolutionDocument8 pagesSérie #3 - S4 2021 - Solutionharedwaiss2010Pas encore d'évaluation
- Cours - Chimie - Dosage - Acide - Base - Bac Toutes Sections (2019-2020) MR Sfaxi SalahDocument3 pagesCours - Chimie - Dosage - Acide - Base - Bac Toutes Sections (2019-2020) MR Sfaxi Salahgzqg9cp7mtPas encore d'évaluation
- Cours3 ElectroniqueI 0506Document7 pagesCours3 ElectroniqueI 0506HALAILIPas encore d'évaluation
- 12 Formulaire 20des 20poutresDocument4 pages12 Formulaire 20des 20poutresyassine lebiouiPas encore d'évaluation
- Chapitre5 Conversion DenergieDocument31 pagesChapitre5 Conversion Denergiehacinemohamed451Pas encore d'évaluation
- Le Premier Principe de La ThermodynamiquDocument2 pagesLe Premier Principe de La ThermodynamiquLeila BachiriPas encore d'évaluation
- Van Der Waals PDFDocument5 pagesVan Der Waals PDFazzamPas encore d'évaluation
- Van Der Waals PDFDocument5 pagesVan Der Waals PDFBilal Bouakrif100% (1)
- Thermo Chap 4 BenchanaaDocument35 pagesThermo Chap 4 Benchanaaadnan aitlahcPas encore d'évaluation
- TD 1 Gaz ParfaitDocument4 pagesTD 1 Gaz Parfaitالـيــاس 『ÃĞ』Pas encore d'évaluation
- A Equilibres Acide Base Power PDFDocument57 pagesA Equilibres Acide Base Power PDFmenin noorPas encore d'évaluation
- 40-102 Équilibre Chimique PDFDocument2 pages40-102 Équilibre Chimique PDFOmar ErrajiPas encore d'évaluation
- UICI Mécanique Des Fluides Chapitre 5Document6 pagesUICI Mécanique Des Fluides Chapitre 5siele.sekongoPas encore d'évaluation
- Distillation - Rectification Étudiant 2022-2023Document24 pagesDistillation - Rectification Étudiant 2022-2023Dounia BenPas encore d'évaluation
- Mecanique Des Fluides 2016Document27 pagesMecanique Des Fluides 2016saif100% (1)
- Diagramme de PhasesDocument98 pagesDiagramme de Phaseskabli ilyassPas encore d'évaluation
- Chap3 T5 Eleve 2223 PDFDocument40 pagesChap3 T5 Eleve 2223 PDFCHELLALI Rabah Kheir EddinePas encore d'évaluation
- Chapitre Fluide1 Watsapp PDFDocument21 pagesChapitre Fluide1 Watsapp PDFEL MAHDI EL WARADY100% (1)
- Le Mercure Et Ses Ions en Solution Aqueuse AcideDocument5 pagesLe Mercure Et Ses Ions en Solution Aqueuse Acideneval chenchouniPas encore d'évaluation
- DoacidfaDocument3 pagesDoacidfaMOUNA MESFARPas encore d'évaluation
- Applications Equations LocalesDocument13 pagesApplications Equations Localesmeriemmalika.aibPas encore d'évaluation
- TD4 - PHY303 2018 2019 CorrectionsDocument11 pagesTD4 - PHY303 2018 2019 CorrectionsWald BahPas encore d'évaluation
- RAFFINAGEDocument28 pagesRAFFINAGEkeassemon danielle mondesirPas encore d'évaluation
- planPV CDocument3 pagesplanPV CNadia AmiriPas encore d'évaluation
- Corrigé TD1Document3 pagesCorrigé TD1Aytaç AktuğPas encore d'évaluation
- Cours de Thermo n2 DR NitcheuDocument49 pagesCours de Thermo n2 DR NitcheuArnauld LefortPas encore d'évaluation
- Amphi 2Document40 pagesAmphi 2api-3830967Pas encore d'évaluation
- Correction Examen Thermodynamique SN 2019-2020Document4 pagesCorrection Examen Thermodynamique SN 2019-2020Ousan NonPas encore d'évaluation
- Diagrammes D EquilibreDocument36 pagesDiagrammes D EquilibreFrodon SacquetPas encore d'évaluation
- TD MDF L2 GP Manomètre À ClocheDocument11 pagesTD MDF L2 GP Manomètre À ClochemelissaelkfelPas encore d'évaluation
- 12 - Formulaire Des PoutresDocument4 pages12 - Formulaire Des PoutresIsmaëlPas encore d'évaluation
- Chapitre III-Le Premier Principe de La Themodynamique Pour Un Système Fermé PDFDocument13 pagesChapitre III-Le Premier Principe de La Themodynamique Pour Un Système Fermé PDFInes MazgarPas encore d'évaluation
- Chapitre III-Le Premier Principe de La Themodynamique Pour Un Système FerméDocument13 pagesChapitre III-Le Premier Principe de La Themodynamique Pour Un Système FerméHamza DerbaliPas encore d'évaluation
- Chap 2 Thermodynamique GAZ REELSDocument39 pagesChap 2 Thermodynamique GAZ REELSNour El Houda TebbanePas encore d'évaluation
- Distillation 1 - CopieDocument46 pagesDistillation 1 - CopieSimo BoumahrachiPas encore d'évaluation
- Examen National Physique Chimie Sciences Maths 2019 Rattrapage CorrigeDocument8 pagesExamen National Physique Chimie Sciences Maths 2019 Rattrapage CorrigeWahbi AbdollahPas encore d'évaluation
- Machine Hydraulique22 (1) (2) - CompressedDocument121 pagesMachine Hydraulique22 (1) (2) - CompressedOualid CatibPas encore d'évaluation
- Chimie 4Document12 pagesChimie 4riadhPas encore d'évaluation
- BenhaklaDocument7 pagesBenhaklakada hanafiPas encore d'évaluation
- Chap 1 Annexe1Document2 pagesChap 1 Annexe1bsqmwmw77sPas encore d'évaluation
- TD5 ThermoDocument3 pagesTD5 Thermocool clipsPas encore d'évaluation
- CorrectionDocument4 pagesCorrectionNan NoussPas encore d'évaluation
- 201801chimie-Mp - Pt-Hachette - PDF 2Document260 pages201801chimie-Mp - Pt-Hachette - PDF 2Rawda KhPas encore d'évaluation
- Rappel SuitesDocument3 pagesRappel SuitesRawda KhPas encore d'évaluation
- Chimie Organique Fonctionelle S5Document120 pagesChimie Organique Fonctionelle S5fskPas encore d'évaluation
- TD FasciculeDocument35 pagesTD FasciculeRawda Kh100% (2)
- Technique de CommunicationDocument3 pagesTechnique de CommunicationRawda KhPas encore d'évaluation
- 201801chimie MP PT Hachette PDFDocument260 pages201801chimie MP PT Hachette PDFRawda KhPas encore d'évaluation
- Technique de CommunicationDocument3 pagesTechnique de CommunicationRawda KhPas encore d'évaluation
- Solubilité ExosDocument1 pageSolubilité ExosRawda KhPas encore d'évaluation
- Chap 2 Alc ®nes Polyccopi © Sans SCH ©masDocument11 pagesChap 2 Alc ®nes Polyccopi © Sans SCH ©masRawda KhPas encore d'évaluation
- Chapitre 1 - Généralités Sur Les Titrages Volumétriques-ConvertiDocument7 pagesChapitre 1 - Généralités Sur Les Titrages Volumétriques-ConvertiRawda KhPas encore d'évaluation
- Dosage Des Ions CLDocument6 pagesDosage Des Ions CLLarusanPas encore d'évaluation
- 201801chimie MP PT Hachette PDFDocument260 pages201801chimie MP PT Hachette PDFRawda KhPas encore d'évaluation
- SériesDocument14 pagesSériesRawda KhPas encore d'évaluation
- 201801chimie MP PT Hachette PDFDocument260 pages201801chimie MP PT Hachette PDFRawda KhPas encore d'évaluation
- Chapitre 1 - Généralités Sur Les Titrages Volumétriques-ConvertiDocument7 pagesChapitre 1 - Généralités Sur Les Titrages Volumétriques-ConvertiRawda KhPas encore d'évaluation
- Resume NomenclatureDocument4 pagesResume NomenclatureRawda KhPas encore d'évaluation
- SériesDocument14 pagesSériesRawda KhPas encore d'évaluation
- TD FasciculeDocument35 pagesTD FasciculeRawda Kh100% (2)
- Série 2Document1 pageSérie 2Rawda KhPas encore d'évaluation
- Doc1 Melanges Binaires Liq VapDocument12 pagesDoc1 Melanges Binaires Liq VapRawda KhPas encore d'évaluation
- Solubilité ExosDocument1 pageSolubilité ExosRawda KhPas encore d'évaluation
- Acides BasesDocument1 pageAcides BasesRawda KhPas encore d'évaluation
- Rappel SuitesDocument3 pagesRappel SuitesRawda KhPas encore d'évaluation
- Resume NomenclatureDocument4 pagesResume NomenclatureRawda KhPas encore d'évaluation
- TD FasciculeDocument35 pagesTD FasciculeRawda Kh100% (2)
- 01 SlidesDocument53 pages01 SlidesjbouguechalPas encore d'évaluation
- GCH2004 Chap1Document15 pagesGCH2004 Chap1dubequimaoPas encore d'évaluation
- C PT 2020 PDFDocument16 pagesC PT 2020 PDFHammouda ChebbiPas encore d'évaluation
- Application Transfert AbsorptionDocument13 pagesApplication Transfert Absorptionben yahya ezzeddinePas encore d'évaluation
- Liquefaction D'un GazDocument21 pagesLiquefaction D'un GazKHALEDFEKAIRPas encore d'évaluation
- Loi de Raoult Et DistillationDocument8 pagesLoi de Raoult Et DistillationChristy Gross100% (1)
- H Prépa Chimie Exercices Et Problèmes MP by André Durupthy, Odile Durupthy, Alain JaubertDocument210 pagesH Prépa Chimie Exercices Et Problèmes MP by André Durupthy, Odile Durupthy, Alain JaubertAngela100% (1)
- Poly 13x14 BinairesDocument33 pagesPoly 13x14 BinairesMohammed Az-GOUNDERPas encore d'évaluation
- TP Miscib P DahDocument8 pagesTP Miscib P DahMustapha Keddous100% (1)
- DistillationExtraction V1Document181 pagesDistillationExtraction V1Hicham El BazidiPas encore d'évaluation
- Cours S4 - VIDocument16 pagesCours S4 - VIEmadPas encore d'évaluation
- Cours Système Binaire Liquide-VapeurDocument60 pagesCours Système Binaire Liquide-VapeurYounes Nikki AlouaniPas encore d'évaluation
- Polycopie Simulateurs Des Procedes Pour Licence GPDocument23 pagesPolycopie Simulateurs Des Procedes Pour Licence GPpc takouachetPas encore d'évaluation
- DistillationExtraction V1Document181 pagesDistillationExtraction V1wafa100% (1)
- DistillationExtraction V1Document181 pagesDistillationExtraction V1Samuel Nikhil Avaby100% (1)
- CHap-II Solutions PropriétésDocument48 pagesCHap-II Solutions PropriétésDô FlamîîngoPas encore d'évaluation
- PVT 2Document9 pagesPVT 2Dja LilPas encore d'évaluation
- Absorption ProfDocument14 pagesAbsorption ProfNouha Jhider80% (5)
- Expérience 2: Purification de Produits Chimiques Par DistillationDocument15 pagesExpérience 2: Purification de Produits Chimiques Par DistillationCassandra MaaloufPas encore d'évaluation
- Poly 13x14 Binaires LV 1ère Partie Miscibilité À Létat Liquide PDFDocument27 pagesPoly 13x14 Binaires LV 1ère Partie Miscibilité À Létat Liquide PDFPaul DayangPas encore d'évaluation
- Dist Chap1Document15 pagesDist Chap1henry0% (1)
- Gouttes 1Document200 pagesGouttes 1dantonkuPas encore d'évaluation
- Cours Thermo - Chapitre 7. Solutions Réelles - 2019Document27 pagesCours Thermo - Chapitre 7. Solutions Réelles - 2019ait hssainPas encore d'évaluation
- Chapitre 5. Solutions IdéalesDocument19 pagesChapitre 5. Solutions Idéaleshayat LOUKILIPas encore d'évaluation
- CHAPITRE2 Distillation Licence 2019-2020 PDFDocument2 pagesCHAPITRE2 Distillation Licence 2019-2020 PDFYasmine Hdjm100% (1)
- Travaux Dirigés de Distillation-ConvertiDocument4 pagesTravaux Dirigés de Distillation-ConvertiSaâd Sardi100% (1)
- Opérations UnitairesDocument16 pagesOpérations Unitaireshadi dzPas encore d'évaluation