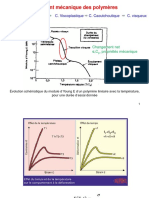
Vous aimerez peut-être aussi
- Strategor 2019 CompletDocument708 pagesStrategor 2019 CompletOumaima Temara100% (9)
- BTS CPI 1 CH 7 PolymèresDocument6 pagesBTS CPI 1 CH 7 PolymèresLucas Filipuzzi100% (2)
- Exercices-Materiaux-Chapitre IDocument8 pagesExercices-Materiaux-Chapitre IJewel Slam100% (1)
- TD Polymere TcaDocument6 pagesTD Polymere TcaBihina HamanPas encore d'évaluation
- TP Polymère + Compte Rendu - TP1 - Identification Des Polymères 6510Document9 pagesTP Polymère + Compte Rendu - TP1 - Identification Des Polymères 6510ahmed ahmadPas encore d'évaluation
- TP Polymère + Compte Rendu - TP2 - Observation Des Sphérolites Au Microscopes Optique en Lumière Polarisée 6511Document3 pagesTP Polymère + Compte Rendu - TP2 - Observation Des Sphérolites Au Microscopes Optique en Lumière Polarisée 6511ahmed ahmadPas encore d'évaluation
- Exercicess Grammaire Explique Du FrancaisDocument224 pagesExercicess Grammaire Explique Du FrancaisAnonymous lg8UvHK100% (12)
- Chapitre I - M1Document14 pagesChapitre I - M1Ayôub ZnPas encore d'évaluation
- Interac MolecDocument2 pagesInterac Molecrachchem9586Pas encore d'évaluation
- À Apprendre Par Coeur 1Document4 pagesÀ Apprendre Par Coeur 1Awatif BePas encore d'évaluation
- Les ThermoplastiquesDocument1 pageLes ThermoplastiquesAhmed AslaouiPas encore d'évaluation
- Chapitre 5 Propriétés Des PolymèresDocument5 pagesChapitre 5 Propriétés Des PolymèresItachi AMVPas encore d'évaluation
- Chapitre 2 Techniques D Analyse Des PolymeresDocument18 pagesChapitre 2 Techniques D Analyse Des PolymeresTunENSTABPas encore d'évaluation
- Chapitre 2 Cristallisation Et FusionDocument38 pagesChapitre 2 Cristallisation Et FusionhadjerPas encore d'évaluation
- ArticleDocument3 pagesArticleKHALI AMIPas encore d'évaluation
- Cours 2 PolyDocument24 pagesCours 2 PolyHoussem HassanetPas encore d'évaluation
- Les Polymères de ThermoformageDocument10 pagesLes Polymères de ThermoformageJay SlvatrPas encore d'évaluation
- Polymer eDocument6 pagesPolymer eMathis LangevinPas encore d'évaluation
- En12 1Document2 pagesEn12 1eljani aminPas encore d'évaluation
- Cor12 1Document2 pagesCor12 1Sou Lef JellaliPas encore d'évaluation
- 2011 353 354 Juin Juil Aout p.74 Cramail HDDocument9 pages2011 353 354 Juin Juil Aout p.74 Cramail HDMebarka TimPas encore d'évaluation
- Chimie Des PolymèresDocument36 pagesChimie Des PolymèresRazvy100% (2)
- Examen Rhologie Et Mise en Uvre Des PolymresDocument3 pagesExamen Rhologie Et Mise en Uvre Des PolymresKha LidPas encore d'évaluation
- Transition Des PolymèresDocument28 pagesTransition Des Polymèresuiopkksfe0% (1)
- Injection CoursDocument10 pagesInjection Coursfranck rogerPas encore d'évaluation
- tp4 Influence Des Traitements Thermiques Sur Les Propric3a9tc3a9s Mc3a9caniques Des Polymc3a8res - Duretc3a9 Et Rc3a9silienceDocument11 pagestp4 Influence Des Traitements Thermiques Sur Les Propric3a9tc3a9s Mc3a9caniques Des Polymc3a8res - Duretc3a9 Et Rc3a9silienceAnoir MatiPas encore d'évaluation
- TP PolymèresDocument9 pagesTP PolymèresTiti Rahim0% (1)
- Comportement Mécanique Des Polymères Partie ADocument9 pagesComportement Mécanique Des Polymères Partie Aali BourenanePas encore d'évaluation
- Table Des Matières: 2 Rhéologie 9Document14 pagesTable Des Matières: 2 Rhéologie 9Mario StiflerPas encore d'évaluation
- Sujet Master 2018Document22 pagesSujet Master 2018dallagi mohamedPas encore d'évaluation
- Cours ThermodynamiqueDocument36 pagesCours ThermodynamiqueMeryem RakbaPas encore d'évaluation
- TP CPG PDFDocument11 pagesTP CPG PDFnadpharm13Pas encore d'évaluation
- Thermodynamique Mpsi NathanDocument157 pagesThermodynamique Mpsi NathanJunior MekavingPas encore d'évaluation
- Modelisation Numerique Des Phenomenes de ChauffageDocument10 pagesModelisation Numerique Des Phenomenes de ChauffageSimo El joumailiPas encore d'évaluation
- Chapitre 33 Structure Comportement Des PolymeresDocument21 pagesChapitre 33 Structure Comportement Des PolymeresCHERIFPas encore d'évaluation
- Membranes NanoDocument9 pagesMembranes NanoOliver FrerePas encore d'évaluation
- CFM2007 0400Document7 pagesCFM2007 0400aminePas encore d'évaluation
- La PlasturgieDocument166 pagesLa Plasturgiemarc schillingPas encore d'évaluation
- Chapitre II Méthodes D'analysesx PDFDocument29 pagesChapitre II Méthodes D'analysesx PDFVerginica SpilosomaPas encore d'évaluation
- Afm GeneralitesDocument6 pagesAfm GeneralitesKhouloud Benkara MedPas encore d'évaluation
- Corr M1 Génie Des Mater Module Prpriétés Physicochimique Des PolymèresDocument4 pagesCorr M1 Génie Des Mater Module Prpriétés Physicochimique Des PolymèresYetd ToudiPas encore d'évaluation
- Polycopié de TP Cinétique 2021 2022Document18 pagesPolycopié de TP Cinétique 2021 2022Yasmine OuabibiPas encore d'évaluation
- Journal Homepage: - : IntroductionDocument14 pagesJournal Homepage: - : IntroductionIJAR JOURNALPas encore d'évaluation
- Vieillissement Des Polymères 2Document9 pagesVieillissement Des Polymères 2ass mohPas encore d'évaluation
- Transferts TermiquesDocument22 pagesTransferts TermiquesRamzi SmidaPas encore d'évaluation
- 4 Proprietes Des MacromleculesDocument4 pages4 Proprietes Des Macromleculescristallisation100% (1)
- Quelle Énergie Pour Demain 12345Document25 pagesQuelle Énergie Pour Demain 12345Louah SafaePas encore d'évaluation
- NanomateriauxDocument29 pagesNanomateriauxKenza HatrounePas encore d'évaluation
- Refdp 201441 P 18Document5 pagesRefdp 201441 P 18anis bouaddouPas encore d'évaluation
- TD Matériaux GC-L2 Manefouet Kentsa-Mai 2021 FlattenDocument3 pagesTD Matériaux GC-L2 Manefouet Kentsa-Mai 2021 FlattenFranck BitaPas encore d'évaluation
- Chapitre 2Document3 pagesChapitre 2Zaki ChekirebPas encore d'évaluation
- Température de Transition VitreuseDocument7 pagesTempérature de Transition VitreuseFrederic Wust100% (1)
- Refdp 20088 P 14Document3 pagesRefdp 20088 P 14Aziz TAOUFYQPas encore d'évaluation
- Notion de Resistance Thermique 223146Document5 pagesNotion de Resistance Thermique 223146Soufiane OuassouPas encore d'évaluation
- Thermo PlastiqueDocument8 pagesThermo PlastiqueFrederic WustPas encore d'évaluation
- Polymères ThermoplastiquesDocument8 pagesPolymères ThermoplastiquesAbderrahim BelmJouJ100% (1)
- CIFA BucharestROMANIA2008Document7 pagesCIFA BucharestROMANIA2008Malika AchouriPas encore d'évaluation
- Processus irréversibles non linéaires en thermodynamique: Les Grands Articles d'UniversalisD'EverandProcessus irréversibles non linéaires en thermodynamique: Les Grands Articles d'UniversalisPas encore d'évaluation
- Énergie thermonucléaire: Les Grands Articles d'UniversalisD'EverandÉnergie thermonucléaire: Les Grands Articles d'UniversalisPas encore d'évaluation
- 3 La Parabole Du SemeurDocument7 pages3 La Parabole Du SemeurIsabela VasconceloPas encore d'évaluation
- TD MicroprocesseurDocument6 pagesTD MicroprocesseurtchoudjafandjaPas encore d'évaluation
- Le Monde Diplomatique 2014 12Document28 pagesLe Monde Diplomatique 2014 12SebastienHoratioKialikiPas encore d'évaluation
- Aide Mémoire - 2023 - SSTDocument31 pagesAide Mémoire - 2023 - SSTJunior METOGO100% (1)
- SAB Mocaf AF2 - AjourDocument31 pagesSAB Mocaf AF2 - AjourCaleb YabadaPas encore d'évaluation
- CV 2 Pages Houcem Eddin MECHICHIDocument2 pagesCV 2 Pages Houcem Eddin MECHICHIHoussem MechichiPas encore d'évaluation
- Jonnesway Outillage-CatalogoFR2017.compressed PDFDocument170 pagesJonnesway Outillage-CatalogoFR2017.compressed PDFconstantine.cherifPas encore d'évaluation
- Avis Bon CommandeDocument4 pagesAvis Bon Commandezak.sb22Pas encore d'évaluation
- LoveDocument4 pagesLoveassadabdi71Pas encore d'évaluation
- Sec 52 QsgengDocument29 pagesSec 52 Qsgengtaiwa1 1Pas encore d'évaluation
- Consultationprojet SIRHDocument25 pagesConsultationprojet SIRHMami cisséPas encore d'évaluation
- MANDATS Gonaives Coopératives Scolaires 315 318 319 320Document4 pagesMANDATS Gonaives Coopératives Scolaires 315 318 319 320SaidEtebbaiPas encore d'évaluation
- rajaonarisonNirinaR ESPA MAST 21 SPAHDocument96 pagesrajaonarisonNirinaR ESPA MAST 21 SPAHRatovoarisoaPas encore d'évaluation
- Conférences - Visites Novembre 2017 PanDocument2 pagesConférences - Visites Novembre 2017 PanPierre-Alexandre NICOLASPas encore d'évaluation
- Pillet SOLEIL 2014Document97 pagesPillet SOLEIL 2014SIAKA OuattaraPas encore d'évaluation
- ElectionDocument58 pagesElectionSerigne Saliou DionguePas encore d'évaluation
- Section 1Document3 pagesSection 1Imane MegzariPas encore d'évaluation
- Chapitre III: Transmittance Ou Fonction de Transfert D'un Système LinéaireDocument4 pagesChapitre III: Transmittance Ou Fonction de Transfert D'un Système LinéaireBouzid Mohamed CherifPas encore d'évaluation
- Acidocétose Diabétique EMC 2018Document11 pagesAcidocétose Diabétique EMC 2018chemmiPas encore d'évaluation
- Attestation Droit SDocument2 pagesAttestation Droit SAchraf FaddouliPas encore d'évaluation
- Reseaux HT TDDocument6 pagesReseaux HT TDPUMA100% (1)
- Manuel D'utilisation Du GEVASCODocument18 pagesManuel D'utilisation Du GEVASCOHervé HollardPas encore d'évaluation
- DAF XF Brochure 2016 FR 67494Document32 pagesDAF XF Brochure 2016 FR 67494Abdel Taleb100% (1)
- AMI Etudes Urbaines Reste Tranche 2 VER DefDocument10 pagesAMI Etudes Urbaines Reste Tranche 2 VER DefAhmed BounenniPas encore d'évaluation
- Exercice 1:: Partie I: Les Réponses Aux Questions Des Deux Exercices Sont ObligatoiresDocument2 pagesExercice 1:: Partie I: Les Réponses Aux Questions Des Deux Exercices Sont ObligatoiresElisa Ger100% (1)
- Protheses de Membre SuperieurDocument19 pagesProtheses de Membre SuperieurAyoub HbPas encore d'évaluation
- Robert Elger Les Nouveaux PotagersDocument195 pagesRobert Elger Les Nouveaux PotagersAmeliePas encore d'évaluation
- E I RachialgiesDocument20 pagesE I RachialgiesMohamed Aziz BaltiPas encore d'évaluation