Vous aimerez peut-être aussi
- Introduction À La Science Des Matériaux - Les Polymères - Propriétés Générales - WikiversitéDocument13 pagesIntroduction À La Science Des Matériaux - Les Polymères - Propriétés Générales - WikiversitéLaurent MorgePas encore d'évaluation
- Conversion De L'Énergie Thermique Des Océans: Des différences de température entre les eaux de surface et les eaux profondes de l'océanD'EverandConversion De L'Énergie Thermique Des Océans: Des différences de température entre les eaux de surface et les eaux profondes de l'océanPas encore d'évaluation
- Cours Polymères-1Document22 pagesCours Polymères-1Ouehnia tynaPas encore d'évaluation
- 1.polymérisation RadicalaireDocument36 pages1.polymérisation Radicalaireomatr0% (1)
- Introduction Polymeres CompositesDocument48 pagesIntroduction Polymeres Compositescdm22orange100% (1)
- Synthèse 6Document22 pagesSynthèse 6Jeyani JEYARAJAHPas encore d'évaluation
- Ingénierie Des POLYMERESDocument70 pagesIngénierie Des POLYMERESAbdel Oihab100% (1)
- Chapitre I POLYMERE INTRODUCTIONDocument43 pagesChapitre I POLYMERE INTRODUCTIONEya B. TalebPas encore d'évaluation
- Lahmaza Ibtissam PDFDocument86 pagesLahmaza Ibtissam PDFAs maPas encore d'évaluation
- Les PolymeresDocument17 pagesLes PolymeresChouaib Aribi100% (1)
- Chap. 1-PolymèresDocument5 pagesChap. 1-PolymèresBénédicte & Christophe MULLERPas encore d'évaluation
- Chapitre 5 Propriétés Des PolymèresDocument5 pagesChapitre 5 Propriétés Des PolymèresItachi AMVPas encore d'évaluation
- Chapitre IiDocument12 pagesChapitre IiIs Lam EdPas encore d'évaluation
- Comportement Mécanique Des Matériaux Polymères Chapitre I M1 S2 GM D.bedgHIOUDocument25 pagesComportement Mécanique Des Matériaux Polymères Chapitre I M1 S2 GM D.bedgHIOUBoubou FouadPas encore d'évaluation
- PrécisDocument34 pagesPrécisArmandPas encore d'évaluation
- Fabrication Du PolypropylèneDocument17 pagesFabrication Du PolypropylèneMoha MidouPas encore d'évaluation
- Mémoire AssassiDocument74 pagesMémoire AssassiYc YacinePas encore d'évaluation
- BoussakDocument175 pagesBoussakMohamed BelbarakaPas encore d'évaluation
- PolypropylèneDocument10 pagesPolypropylènejosué mwambaPas encore d'évaluation
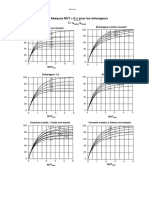
- Abaques NUT F (E)Document1 pageAbaques NUT F (E)aberranePas encore d'évaluation
- Charbon Actif PorositéDocument17 pagesCharbon Actif PorositéFay Rouz Taleb EpTahri100% (1)
- Polymerisations IoniquesDocument60 pagesPolymerisations IoniquesAd ElPas encore d'évaluation
- Monographie PolymèresDocument32 pagesMonographie Polymèresilio6Pas encore d'évaluation
- Hamadouche Hanane Et Bouarab NaceraDocument57 pagesHamadouche Hanane Et Bouarab NaceraHALIMAPas encore d'évaluation
- Calcul de Vérification de La Colonne de Distillation de L'unité Topping de La Raffinerie de Hassi MessaoudDocument146 pagesCalcul de Vérification de La Colonne de Distillation de L'unité Topping de La Raffinerie de Hassi Messaoudtrc.berlinPas encore d'évaluation
- La Technologie Du BetonDocument111 pagesLa Technologie Du Betonعلي ابو خالدPas encore d'évaluation
- Chapitre 2 PDFDocument10 pagesChapitre 2 PDFHadjer ZitounePas encore d'évaluation
- Structures Conformations PolyDocument51 pagesStructures Conformations Polyanon_509931202Pas encore d'évaluation
- Vieillissement Des PolymèresDocument11 pagesVieillissement Des PolymèresHamza BEYADPas encore d'évaluation
- Température de Transition VitreuseDocument7 pagesTempérature de Transition VitreuseFrederic Wust100% (1)
- Cours VapocraquageDocument7 pagesCours VapocraquageHä'deel ÖdenmemişPas encore d'évaluation
- MANUELDocument130 pagesMANUELHadjer ZitounePas encore d'évaluation
- Memoire de Master Sur La CorrosionDocument65 pagesMemoire de Master Sur La CorrosionAbir siliniPas encore d'évaluation
- Methodes D'elaborationDocument12 pagesMethodes D'elaborationS0UM0H100% (1)
- Rappels Thermo Des Solutions MacromoléculairesDocument4 pagesRappels Thermo Des Solutions MacromoléculairesVincent ImadPas encore d'évaluation
- Chimie VerteDocument28 pagesChimie VerteWa SsimPas encore d'évaluation
- Mémoire Finale m2 Aib Rahma&Khorief Nacer Eddine ChaimaDocument98 pagesMémoire Finale m2 Aib Rahma&Khorief Nacer Eddine ChaimaRãhmã Râ Hma0% (1)
- Mémoire Complet - pdf1Document76 pagesMémoire Complet - pdf1Ouehnia tynaPas encore d'évaluation
- Les PolymèresDocument36 pagesLes PolymèresOussama El BouadiPas encore d'évaluation
- Master Electrochimie Et Corrosion PDFDocument26 pagesMaster Electrochimie Et Corrosion PDFFati fatiPas encore d'évaluation
- These DoumbouyaDocument119 pagesThese DoumbouyaMohamed ELMOUHINNIPas encore d'évaluation
- Séance6 Réacteurs Industriels 20-21Document86 pagesSéance6 Réacteurs Industriels 20-2105-BENHAIBA WAFAEPas encore d'évaluation
- VaposDocument20 pagesVaposJESuisAhmedPas encore d'évaluation
- Généralités Sur Les PolymèresDocument5 pagesGénéralités Sur Les PolymèresHaithem RamoulPas encore d'évaluation
- Exposé de PolystyréneDocument12 pagesExposé de PolystyréneAn Gel100% (1)
- Polycopie de Cours Thermodynamique Premiere AnneeDocument89 pagesPolycopie de Cours Thermodynamique Premiere AnneejouPas encore d'évaluation
- Mémoire Finale LADJAL PDFDocument146 pagesMémoire Finale LADJAL PDFGhada19 TiahPas encore d'évaluation
- 3A BT MCS - Matières PlastiquesDocument6 pages3A BT MCS - Matières PlastiquesFélix KouassiPas encore d'évaluation
- Contribution A Letude de Linhibition de Corrosion Dun AcierDocument50 pagesContribution A Letude de Linhibition de Corrosion Dun AcierterPas encore d'évaluation
- Procédé de Fabrication Du PolypropylèneDocument16 pagesProcédé de Fabrication Du PolypropylèneMoha MidouPas encore d'évaluation
- Cours PolymèresDocument17 pagesCours PolymèresJhee raaPas encore d'évaluation
- These de Doctorat Es-Science Final Et Modifier 08-07-2017Document208 pagesThese de Doctorat Es-Science Final Et Modifier 08-07-2017Hamza El MrabetPas encore d'évaluation
- Oussad, AliDocument71 pagesOussad, AliAmir NasserPas encore d'évaluation
- Futur Ceramiques PDFDocument15 pagesFutur Ceramiques PDFFida ForkanePas encore d'évaluation
- TH3094Document101 pagesTH3094wissoubenaouda2110Pas encore d'évaluation
- Cours Poly. 2023 Etudiant (E) SDocument57 pagesCours Poly. 2023 Etudiant (E) SZakaria AlouanePas encore d'évaluation
- Examen 202O Cours Céramique Ilham ElbaraiDocument8 pagesExamen 202O Cours Céramique Ilham ElbaraiNouhaila HamroudPas encore d'évaluation
- Etude Par Spectroscopie Raman Et Modelisation Dune Resine Composite RTM PDFDocument136 pagesEtude Par Spectroscopie Raman Et Modelisation Dune Resine Composite RTM PDFKhaLed BenKaPas encore d'évaluation
- Serie 1 MC 23-24Document2 pagesSerie 1 MC 23-24Adel Hammia Bn AliPas encore d'évaluation
- Partie 2 PolyméresDocument67 pagesPartie 2 PolyméresOns KammounPas encore d'évaluation
- Applications de la spectrophotomérie en phytochimie: sciencesD'EverandApplications de la spectrophotomérie en phytochimie: sciencesPas encore d'évaluation
- Évolution des procédés - la séparation de l'air atmosphérique en ses éléments, l'oxygène et l'azoteD'EverandÉvolution des procédés - la séparation de l'air atmosphérique en ses éléments, l'oxygène et l'azotePas encore d'évaluation
- Le Guide de La Sécurité Au Travail by Benoît PéribèreDocument1 pageLe Guide de La Sécurité Au Travail by Benoît PéribèreHamid BouleghabPas encore d'évaluation
- EduQua Manuel 121Document46 pagesEduQua Manuel 121Hamid BouleghabPas encore d'évaluation
- Auto Diagnostic ManagerDocument5 pagesAuto Diagnostic ManagerHamid BouleghabPas encore d'évaluation
- Gestion Des Risques 2e EditionDocument1 pageGestion Des Risques 2e EditionHamid BouleghabPas encore d'évaluation
- InraDocument16 pagesInraHamid BouleghabPas encore d'évaluation
- Protocole PL v12b 03022014 079483700 0926 03032014Document154 pagesProtocole PL v12b 03022014 079483700 0926 03032014Hamid BouleghabPas encore d'évaluation
- L Evolution de La Qualite 2Document13 pagesL Evolution de La Qualite 2Hamid Bouleghab100% (1)
- Les Quatre Niveaux Du Management Des Compétences: Individuel, Collectif, Stratégique Et EnvironnementalDocument19 pagesLes Quatre Niveaux Du Management Des Compétences: Individuel, Collectif, Stratégique Et EnvironnementalHamid BouleghabPas encore d'évaluation
- Controle Laitier Protocoles Et Methodes de QualificationDocument6 pagesControle Laitier Protocoles Et Methodes de QualificationHamid BouleghabPas encore d'évaluation
- Abbas Mohamed Larbi Le Management Des Competences Dune Entreprise AlgerinneDocument198 pagesAbbas Mohamed Larbi Le Management Des Competences Dune Entreprise AlgerinneHamid BouleghabPas encore d'évaluation
- Proces Verbal de ReunionDocument1 pageProces Verbal de ReunionHamid BouleghabPas encore d'évaluation
- Méthodologie de Controle Cours de Pharmacie Industrielle 5eme Année Pharmacie DR KARAAR - 2Document7 pagesMéthodologie de Controle Cours de Pharmacie Industrielle 5eme Année Pharmacie DR KARAAR - 2Hamid BouleghabPas encore d'évaluation
- Referent I El 2008Document165 pagesReferent I El 2008Hamid BouleghabPas encore d'évaluation
- Demande Du Code Secret Compte CCPDocument2 pagesDemande Du Code Secret Compte CCPKhaledKhaloudi100% (3)
- Referentiel Des Competences IntergenerationnellesDocument40 pagesReferentiel Des Competences IntergenerationnellesHamid BouleghabPas encore d'évaluation
- Analyse Par Les Dix M Milieu Moment . Main D'œuvre Moyens Méthodes Matières Management Mission Maintenance MesuresDocument1 pageAnalyse Par Les Dix M Milieu Moment . Main D'œuvre Moyens Méthodes Matières Management Mission Maintenance MesuresHamid BouleghabPas encore d'évaluation
- Compte RenduDocument1 pageCompte RenduHamid BouleghabPas encore d'évaluation
- Proces Verbal de ReunionDocument1 pageProces Verbal de ReunionHamid BouleghabPas encore d'évaluation
- Physique Des Polymères PDFDocument30 pagesPhysique Des Polymères PDFMohammed Bouchelarm100% (1)
- Chapitre 0 PROPRIETES DES MATIERES PLASTIQUESDocument49 pagesChapitre 0 PROPRIETES DES MATIERES PLASTIQUESEya B. TalebPas encore d'évaluation
- Chapitre I Matériaux PolymèresDocument9 pagesChapitre I Matériaux Polymèresimene.benyahiaPas encore d'évaluation
- Chap 2Document17 pagesChap 2Hana Salah AiechPas encore d'évaluation
- BtsmacroDocument12 pagesBtsmacrofafoulolPas encore d'évaluation
- Rapport D - Expose Chimie Industrielle PlastiqueDocument46 pagesRapport D - Expose Chimie Industrielle PlastiqueAbderrahim BelmJouJPas encore d'évaluation
- 2022 2023 Matériaux Et Propriétés 1 Généralité 1Document165 pages2022 2023 Matériaux Et Propriétés 1 Généralité 1Wa Ssim50% (2)
- Polymeres 001Document10 pagesPolymeres 001Mohamed HassaniPas encore d'évaluation
- Mini Projet MatériauxDocument32 pagesMini Projet MatériauxLot FiPas encore d'évaluation
- MatériauxII-chapitre1 PolymèreDocument26 pagesMatériauxII-chapitre1 PolymèreHoussem HassanetPas encore d'évaluation
- Materiaux Polymères PDFDocument38 pagesMateriaux Polymères PDFDi SecrètePas encore d'évaluation
- Partie 2 PolyméresDocument67 pagesPartie 2 PolyméresOns KammounPas encore d'évaluation
- MTR1 Chapitre 3 POLYMERES 2023 2024Document8 pagesMTR1 Chapitre 3 POLYMERES 2023 2024chaimabenchikhlehoucinePas encore d'évaluation
- Chapitre I. Structure Des Polymeres Et Leurs ClassificationsDocument8 pagesChapitre I. Structure Des Polymeres Et Leurs ClassificationsJewel SlamPas encore d'évaluation
- TD Polymères 1 Masses Moléculaires Moyennes, Indice de PolydispersitéDocument2 pagesTD Polymères 1 Masses Moléculaires Moyennes, Indice de PolydispersitéHana Salah AiechPas encore d'évaluation
- Encapsulation de Molécules Biologiquement Actives Dans Des Systèmes Complexes À Base de BiopolymèresDocument96 pagesEncapsulation de Molécules Biologiquement Actives Dans Des Systèmes Complexes À Base de Biopolymèresdoha DhohaPas encore d'évaluation
- PFE Finale .Bchir Abdelkrim.99666231Document65 pagesPFE Finale .Bchir Abdelkrim.99666231Karim BchirPas encore d'évaluation
- PolymeresDocument8 pagesPolymeresCAPRIROCKPas encore d'évaluation
- Techno 2 AnneeDocument157 pagesTechno 2 Anneeaidaradaouda11Pas encore d'évaluation
- Chapitre 01Document11 pagesChapitre 01Taim KhouriPas encore d'évaluation
- Cours PDFDocument58 pagesCours PDFAmani bouzaouitPas encore d'évaluation
- MP10 Formation CnamDocument77 pagesMP10 Formation CnamKenya GomezPas encore d'évaluation
- Chapitre III Polymérisation AnioniqueDocument8 pagesChapitre III Polymérisation AnioniqueKherfi CherifaPas encore d'évaluation
- 210-U-10 Polymères Et Résines CompositesDocument26 pages210-U-10 Polymères Et Résines CompositesBESSAIH RymPas encore d'évaluation
- Effet de L'environnement Sur Les Propretés Mécanique Des Tubes de HDPE 2Document70 pagesEffet de L'environnement Sur Les Propretés Mécanique Des Tubes de HDPE 2ahmoudaPas encore d'évaluation
- Polymère GénéralDocument28 pagesPolymère GénéralDaniel LiePas encore d'évaluation
- M1 Polymeres Cours1Document35 pagesM1 Polymeres Cours1peutilouis100% (1)