Vous aimerez peut-être aussi
- ALCENESDocument8 pagesALCENESHadjer zitPas encore d'évaluation
- AlcèneDocument6 pagesAlcènechristophe100% (1)
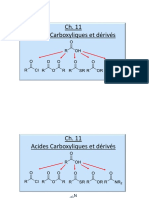
- Acide Carboxyliques (Cours) (2) - ConvertiDocument49 pagesAcide Carboxyliques (Cours) (2) - ConvertiMALLOUK .MOUTAOUAKILPas encore d'évaluation
- 11a - Interactions IntermoleculairesDocument26 pages11a - Interactions Intermoleculairesmariakassis8100% (1)
- Série Dexercices Sur Lélectrode Normale À HydrogèneDocument4 pagesSérie Dexercices Sur Lélectrode Normale À HydrogènechadaPas encore d'évaluation
- Reactivité Chimique - Chapitre 1Document44 pagesReactivité Chimique - Chapitre 1abraham LincolnPas encore d'évaluation
- Potentiel ChimiqueDocument1 pagePotentiel ChimiqueJob ElsonPas encore d'évaluation
- Substitutions EliminationsDocument65 pagesSubstitutions EliminationsYounes MAZOUARPas encore d'évaluation
- Chimie OrganiqueDocument29 pagesChimie OrganiqueHamza SidhoumPas encore d'évaluation
- Chap 4 DiènesDocument16 pagesChap 4 DiènesWahab HoubadPas encore d'évaluation
- Cours4 Les Reactions Chimie Organique Part1Document29 pagesCours4 Les Reactions Chimie Organique Part1Kalosoiretrotchgmail.com KalosoPas encore d'évaluation
- Réaction D'estérification Et D'hydrolyse. Contrôle de L'état Finale D'un Système ChimiqueDocument47 pagesRéaction D'estérification Et D'hydrolyse. Contrôle de L'état Finale D'un Système ChimiqueZakari YaePas encore d'évaluation
- Corrigé - Réactions Chimiques + Acide:BaseDocument12 pagesCorrigé - Réactions Chimiques + Acide:BaseThierryPas encore d'évaluation
- StereochimieDocument17 pagesStereochimieDarel NadjieraPas encore d'évaluation
- Cours - Chimie - Les Acides Et Les BasesDocument6 pagesCours - Chimie - Les Acides Et Les BasesDaghsni Said100% (2)
- Chimie Des Solutions - Chapitre 1Document19 pagesChimie Des Solutions - Chapitre 1Aziz DahhaPas encore d'évaluation
- Chap2 SMP5Document14 pagesChap2 SMP5Daoud El CaidPas encore d'évaluation
- Ch11 Acides Esters AmidesDocument56 pagesCh11 Acides Esters AmidesYugi kevin14Pas encore d'évaluation
- 2° Contrôle SMPC S2 2012 2013Document2 pages2° Contrôle SMPC S2 2012 2013lahssane hmidat100% (1)
- Exercice-Liaison Chimique+Corrigé TypeDocument3 pagesExercice-Liaison Chimique+Corrigé TypejessieholdiePas encore d'évaluation
- Série 1 Chimie Generale AU 2020Document1 pageSérie 1 Chimie Generale AU 2020rekik hibaPas encore d'évaluation
- Réactivité en Chimie Organique PDFDocument9 pagesRéactivité en Chimie Organique PDFAchwak BelfadelPas encore d'évaluation
- Chimie OrgaDocument76 pagesChimie OrgaYoussef AouinPas encore d'évaluation
- Nomenclature Des Composés OrganiquesDocument16 pagesNomenclature Des Composés OrganiquesRuroniPas encore d'évaluation
- Chimie Organique Cours Sur Les Acides Carboxyliques Et Leurs DérivésDocument24 pagesChimie Organique Cours Sur Les Acides Carboxyliques Et Leurs DérivésKone Kouwelton100% (1)
- Chapitre I.2 - Alcènes-Alcynes - B. MESSAOUDI - 14-02-2022Document50 pagesChapitre I.2 - Alcènes-Alcynes - B. MESSAOUDI - 14-02-2022Abderrahmane AEPas encore d'évaluation
- Thermochimie Prof BonDocument41 pagesThermochimie Prof BonAime Desire wogouPas encore d'évaluation
- 6 Reactions D Elimination 1Document9 pages6 Reactions D Elimination 1Blondet RomaricPas encore d'évaluation
- Thermodynamique PDFDocument18 pagesThermodynamique PDFKarim MegherfiPas encore d'évaluation
- 2eme Cours de Chimie OrganiqueDocument44 pages2eme Cours de Chimie OrganiqueAnfel BouchairPas encore d'évaluation
- Acide Base TakiDocument4 pagesAcide Base Takiaziz bensaid2Pas encore d'évaluation
- 3-Le Second Principe de La ThermodynamiqueDocument12 pages3-Le Second Principe de La ThermodynamiqueAntes de Partir, A.C.Pas encore d'évaluation
- Tous Les ChapitresDocument85 pagesTous Les ChapitresAnonymous a9sVQWPas encore d'évaluation
- Cours 8Document2 pagesCours 8Beatrice Florin100% (2)
- Fiche Sur LélectrolyseDocument4 pagesFiche Sur LélectrolysechadaPas encore d'évaluation
- Corrige Examen Session NormaleDocument6 pagesCorrige Examen Session NormaleKhlidPas encore d'évaluation
- Chapitre 3 THERMOCHIMIEDocument6 pagesChapitre 3 THERMOCHIMIEhmza14Pas encore d'évaluation
- C111 - Cours (Atomistique) (Chapitre 2)Document5 pagesC111 - Cours (Atomistique) (Chapitre 2)younescheldi119Pas encore d'évaluation
- Poly SV3 Biophysique 2023Document26 pagesPoly SV3 Biophysique 2023CHAIMAE AFIF100% (1)
- TD Atomistique2Document3 pagesTD Atomistique2pitter PitkethlyPas encore d'évaluation
- Chapitre II - Sétéréochimie B. MESSAOUDI - 04!03!2022Document67 pagesChapitre II - Sétéréochimie B. MESSAOUDI - 04!03!2022Abderrahmane AEPas encore d'évaluation
- Les Bases de La Chimie Cours LatexDocument5 pagesLes Bases de La Chimie Cours LatexYoussra mhaouiPas encore d'évaluation
- Alkylation PDFDocument12 pagesAlkylation PDFNeila HasnaouiPas encore d'évaluation
- Chapitre 4 Couleurs Et ImagesDocument8 pagesChapitre 4 Couleurs Et ImagesLqkchanPas encore d'évaluation
- 1 Intermediaires Reactionnels 1Document12 pages1 Intermediaires Reactionnels 1Rachid Guend100% (1)
- A 3 CompetitionDocument3 pagesA 3 CompetitionEmilie BoubinetPas encore d'évaluation
- Chapitre 6 CINETIQUE CHIMIE PART 2Document9 pagesChapitre 6 CINETIQUE CHIMIE PART 2Faklish LoufiPas encore d'évaluation
- Chimie GénéraleDocument175 pagesChimie GénéraleBopePas encore d'évaluation
- Les ConcentrartionsDocument2 pagesLes ConcentrartionsdonoPas encore d'évaluation
- Cours de Chimie Organique Licence 1 Semestre 1Document32 pagesCours de Chimie Organique Licence 1 Semestre 1Balla SangarePas encore d'évaluation
- Examen L1PCSM Octobre 2015-+CorrigéDocument3 pagesExamen L1PCSM Octobre 2015-+CorrigéSerigne Alassane Dieng100% (1)
- Thermo 1Document6 pagesThermo 1Oussama El BouadiPas encore d'évaluation
- Chap2 Aldéhydes Et CétonesDocument34 pagesChap2 Aldéhydes Et CétonesEssemlali Abde SamadPas encore d'évaluation
- Fonctions OxygeneesDocument29 pagesFonctions OxygeneesharoldkossaPas encore d'évaluation
- Série Liaison Chimiques 2èmeDocument8 pagesSérie Liaison Chimiques 2èmehichriolfa81Pas encore d'évaluation
- Cours CHM146 SVT Semaine6Document11 pagesCours CHM146 SVT Semaine6Abalo KpelemouaPas encore d'évaluation
- AlcynesDocument5 pagesAlcynesمريم ياسمينPas encore d'évaluation
- Chimie Orga 2Document44 pagesChimie Orga 2Yeo Ouanan LassinaPas encore d'évaluation
- Chap.4 OXY RED PDFDocument36 pagesChap.4 OXY RED PDFrajaPas encore d'évaluation
- Glucides: Les Grands Articles d'UniversalisD'EverandGlucides: Les Grands Articles d'UniversalisPas encore d'évaluation
- Exercices Corrigés Chimie QuantiqueDocument4 pagesExercices Corrigés Chimie QuantiqueHk Eh100% (3)
- Feuille Quadrillee PlusDocument1 pageFeuille Quadrillee PlusHk EhPas encore d'évaluation
- TD RMN 2020 PDFDocument9 pagesTD RMN 2020 PDFSekou Kone100% (2)
- Les Huiles EssentiellesDocument22 pagesLes Huiles EssentiellesHk EhPas encore d'évaluation
- Presentation Arduino PDFDocument17 pagesPresentation Arduino PDFguelbaoui younessPas encore d'évaluation
- Resonance Magnetique Nucleaire Bidimensionnelle: RMN 2DDocument20 pagesResonance Magnetique Nucleaire Bidimensionnelle: RMN 2DHk Eh100% (2)
- RMN Molecules Organiques PDFDocument162 pagesRMN Molecules Organiques PDFHamza BoulikaPas encore d'évaluation
- LFC3 RMN Chap 1 + ExercciesDocument64 pagesLFC3 RMN Chap 1 + ExercciesHk EhPas encore d'évaluation
- Exercices Corrigés Chimie QuantiqueDocument4 pagesExercices Corrigés Chimie QuantiqueRottina Rossy100% (1)
- Correction Serie N1 Analytique 1Document6 pagesCorrection Serie N1 Analytique 1Hk Eh100% (1)
- Chapitre 2 RMN 13CDocument35 pagesChapitre 2 RMN 13CHk Eh100% (5)
- Compte Rendu Loscilloscope CathodiqueDocument13 pagesCompte Rendu Loscilloscope CathodiqueL'atelier de licornePas encore d'évaluation
- Devoir de Contrôle N°1 (Corrigé) - SC - Physique - 1ère AS (2010-2011) MR AbdessatarDocument4 pagesDevoir de Contrôle N°1 (Corrigé) - SC - Physique - 1ère AS (2010-2011) MR AbdessatarabdelPas encore d'évaluation
- Les Huiles EssentiellesDocument22 pagesLes Huiles EssentiellesHk EhPas encore d'évaluation
- Objectifs PédagogiquesDocument5 pagesObjectifs PédagogiquesHk EhPas encore d'évaluation
- Rép 5 PR Communiquer en FrancaisDocument5 pagesRép 5 PR Communiquer en FrancaischerazerPas encore d'évaluation
- Feuille Quadrillee PlusDocument1 pageFeuille Quadrillee PlusHk EhPas encore d'évaluation
- Alasmar Eliane 2018Document234 pagesAlasmar Eliane 2018Hk EhPas encore d'évaluation
- Exercices-Avec-Correction Info 2emeDocument56 pagesExercices-Avec-Correction Info 2emeraouf50% (2)
- Diagnostic Mod0Document8 pagesDiagnostic Mod0Hk EhPas encore d'évaluation
- InfirmierDocument7 pagesInfirmierHk EhPas encore d'évaluation
- StageDocument14 pagesStageHk Eh75% (4)
- Physique ChimieDocument10 pagesPhysique ChimieHk EhPas encore d'évaluation
- Fiche 5emeDocument250 pagesFiche 5emetata74% (43)
- Éval Oral PrérequiDocument1 pageÉval Oral PrérequiHk EhPas encore d'évaluation
- 538 Dfbaa 52 B 55Document13 pages538 Dfbaa 52 B 55manel0% (1)
- Résultat Écrit Mod0-ADocument1 pageRésultat Écrit Mod0-AHk EhPas encore d'évaluation
- Nanomateraiux NanotechnologiesDocument4 pagesNanomateraiux NanotechnologiesHk EhPas encore d'évaluation
- Les Huiles EssentiellesDocument22 pagesLes Huiles EssentiellesHk EhPas encore d'évaluation
- Bacacier Gamme 2011Document16 pagesBacacier Gamme 2011sqdqsdPas encore d'évaluation
- ChimieDocument74 pagesChimiePaul FatheadPas encore d'évaluation
- Phytoremédiation - WikipédiaDocument22 pagesPhytoremédiation - WikipédiaJunior NdomPas encore d'évaluation
- 2023 Paper 3 MarkDocument25 pages2023 Paper 3 Markmanuelaa.salccbPas encore d'évaluation
- La GlycérineDocument3 pagesLa GlycérineramzibtrPas encore d'évaluation
- TP Licence de Chimie 2022Document8 pagesTP Licence de Chimie 2022Hajar AMENAOUPas encore d'évaluation
- Molecules Organiques Et Squelettes Carbones Modification Du Squelette Carbone Cours 2 4Document5 pagesMolecules Organiques Et Squelettes Carbones Modification Du Squelette Carbone Cours 2 4Ossama SariaPas encore d'évaluation
- Sc389rie 2 Les AlcanesDocument2 pagesSc389rie 2 Les AlcanesDavid MbangPas encore d'évaluation
- Chapitre II Cours de StereochimieDocument98 pagesChapitre II Cours de StereochimieDOBA OLIVIERPas encore d'évaluation
- LES AMIDES MagazinesDocument3 pagesLES AMIDES Magazinesrania50% (2)
- Cours 01Document89 pagesCours 01aliabidi1652Pas encore d'évaluation
- Correction TD Révision PDFDocument7 pagesCorrection TD Révision PDFTwati Ala100% (1)
- 9 - Le BitumeDocument16 pages9 - Le Bitumeamani.arfaoui1991Pas encore d'évaluation
- Cours-Microbio Industr-Ghellai-3emA-convertiDocument41 pagesCours-Microbio Industr-Ghellai-3emA-convertiL'impératrice Britannique100% (1)
- Cours D'éléments D'agricultureDocument54 pagesCours D'éléments D'agricultureMahonde Bayeb ismaila100% (2)
- P94-047 Teneur en Matiere Organique Par CalcinationDocument8 pagesP94-047 Teneur en Matiere Organique Par CalcinationAnis Bouraba75% (4)
- Chapitre 6 HEDocument22 pagesChapitre 6 HEAlexandre Kpangny BéniPas encore d'évaluation
- Developpement Durable - Identifier Les Matières Plastiques Recyclables TERMINALE Version Finalisée PDFDocument17 pagesDeveloppement Durable - Identifier Les Matières Plastiques Recyclables TERMINALE Version Finalisée PDFMohammed LahssainiPas encore d'évaluation
- Copie Final 1Document37 pagesCopie Final 1Ghileb RihabPas encore d'évaluation
- Sds n7330 FRANCEDocument11 pagesSds n7330 FRANCEtimbulPas encore d'évaluation
- TP Opu Bio 1Document9 pagesTP Opu Bio 1Veronika PrymPas encore d'évaluation
- Cours TIAA1 Chapitre1Document27 pagesCours TIAA1 Chapitre1kokinal897Pas encore d'évaluation
- Cours Régulation Et Facteurs Influencant La CatalyseDocument60 pagesCours Régulation Et Facteurs Influencant La CatalyseMOHAMMED ELKHOUADPas encore d'évaluation
- Resumé TP Biochimie Structurale 1ère Année UIRDocument15 pagesResumé TP Biochimie Structurale 1ère Année UIRd2v4y27jk5Pas encore d'évaluation
- 11201224t.pdfm, Nqno Qteriqux Hybrides Bqndou SQ IrqDocument122 pages11201224t.pdfm, Nqno Qteriqux Hybrides Bqndou SQ Irqbandou samiraPas encore d'évaluation
- M2OSB Exam MS 2020 2021Document5 pagesM2OSB Exam MS 2020 2021ely59000asPas encore d'évaluation
- Cahier Des Charge Piment Langues D'oiseaux NigeriaDocument8 pagesCahier Des Charge Piment Langues D'oiseaux NigeriaArlettePas encore d'évaluation
- Metabolisme MusculaireDocument54 pagesMetabolisme MusculaireRania Ben HaddadaPas encore d'évaluation
- Brochure Technologie Et Systemes D Injection FRDocument10 pagesBrochure Technologie Et Systemes D Injection FRAbdou BidaouiPas encore d'évaluation
- Expériences ChimieDocument9 pagesExpériences Chimieami rPas encore d'évaluation