Vous aimerez peut-être aussi
- Cours Chimie Chap III Principe2 ThermodynamiqueDocument5 pagesCours Chimie Chap III Principe2 ThermodynamiqueDoudou AminePas encore d'évaluation
- Solution-TD1 Thermodynamique Des ÉquilibreDocument12 pagesSolution-TD1 Thermodynamique Des ÉquilibreInas DrPas encore d'évaluation
- Corr EXC8 PHeno de TransfertDocument2 pagesCorr EXC8 PHeno de TransfertRim Sossey AlaouiPas encore d'évaluation
- Cours1 Redress MonoDocument10 pagesCours1 Redress MonoMbaye DIEYEPas encore d'évaluation
- Chapitre 3-2emeprin-ConvertiDocument22 pagesChapitre 3-2emeprin-ConvertiAhlem AchourPas encore d'évaluation
- Cours Mecanique Fluides - Chapitres 6 Et 7Document16 pagesCours Mecanique Fluides - Chapitres 6 Et 7chadiPas encore d'évaluation
- Thermo 3Document2 pagesThermo 3Salah Eddine SafyounPas encore d'évaluation
- TPI - Fluides 1Document26 pagesTPI - Fluides 1Yassin BachiouaPas encore d'évaluation
- Chap IV Second Principe de La Thermodynamique - ST - GC - GeDocument21 pagesChap IV Second Principe de La Thermodynamique - ST - GC - GeJean Marc LengePas encore d'évaluation
- Chapitre 2Document27 pagesChapitre 2AbdouPas encore d'évaluation
- TPMCC Auriol Idriss ElliottDocument13 pagesTPMCC Auriol Idriss Elliottoteme Mbella King DuclairPas encore d'évaluation
- INSa ROUEN - Seismic - Lesson02Document41 pagesINSa ROUEN - Seismic - Lesson02christophesandPas encore d'évaluation
- Cours 6 - SimulinkDocument28 pagesCours 6 - SimulinkaminevirtualPas encore d'évaluation
- Corrigé de La Fiche TD N°01Document6 pagesCorrigé de La Fiche TD N°01ben binahPas encore d'évaluation
- Thermodynamique 444Document9 pagesThermodynamique 444yassineredone4Pas encore d'évaluation
- Cours 4 Second principe 2021Document22 pagesCours 4 Second principe 2021leroy makita loubakiPas encore d'évaluation
- PDFDocument1 pagePDFزكرياء بنحيرتPas encore d'évaluation
- MDF1 StatiqueDocument7 pagesMDF1 StatiqueKadiro OJPas encore d'évaluation
- 2e Principe de La ThermoDocument15 pages2e Principe de La ThermoMohamed MohPas encore d'évaluation
- កំណែរ វិញ្ញាសាប្រឡងបញ្ចប់ឆមាសទី១មេកានិច២Document9 pagesកំណែរ វិញ្ញាសាប្រឡងបញ្ចប់ឆមាសទី១មេកានិច២Dim Lify100% (2)
- Chap2 1-Redmono Noncdés ppt-2021Document37 pagesChap2 1-Redmono Noncdés ppt-2021Landry YobouePas encore d'évaluation
- Thermodynamique Chapitre 3 PDFDocument8 pagesThermodynamique Chapitre 3 PDFNahd RamdanePas encore d'évaluation
- DM Elec2mec1 1011-EsDocument4 pagesDM Elec2mec1 1011-EsAmìnè Cheìkh MèdPas encore d'évaluation
- DM1 - 2009-2010-Es 1Document2 pagesDM1 - 2009-2010-Es 1Ihssane AtraouiPas encore d'évaluation
- Chapitre5 ThermophyDocument6 pagesChapitre5 Thermophyyeregej913Pas encore d'évaluation
- Chapitre IVDocument6 pagesChapitre IValicecullend7Pas encore d'évaluation
- Doc. 4Document5 pagesDoc. 4Adje Kponon-eklouPas encore d'évaluation
- Chap4 Deuxième PrincipeDocument9 pagesChap4 Deuxième PrincipeaminealskjzPas encore d'évaluation
- ENGI Test 2 FormulaireDocument2 pagesENGI Test 2 Formulaireanthony duboisPas encore d'évaluation
- Vibration Exo CorrigerDocument2 pagesVibration Exo CorrigerdjamilPas encore d'évaluation
- Correction TD2 PDFDocument4 pagesCorrection TD2 PDFSamiya CPas encore d'évaluation
- Corrigé TD 3 MarakechDocument5 pagesCorrigé TD 3 MarakechIsmail SalamaPas encore d'évaluation
- Chap 2 La Conduction ThermiqueDocument7 pagesChap 2 La Conduction ThermiqueYosra Jbeli100% (3)
- BoussinesqDocument17 pagesBoussinesqhassanPas encore d'évaluation
- CHAPITRE IV 2eme Principe de La Thermodynamique PDFDocument7 pagesCHAPITRE IV 2eme Principe de La Thermodynamique PDFAsmahane FaslaPas encore d'évaluation
- Chapitre 2 Thermodynamique PhysiqueDocument11 pagesChapitre 2 Thermodynamique PhysiqueChaka TraorePas encore d'évaluation
- Résumé Chapitre 2Document4 pagesRésumé Chapitre 2reguiegyassine098Pas encore d'évaluation
- Examen MDF2 2021Document4 pagesExamen MDF2 2021abdousetif39Pas encore d'évaluation
- Thermo RésuméDocument10 pagesThermo RésuméAmine HamaouiPas encore d'évaluation
- Calcul Des Déformations Par Les Méthodes Énergétiques (CASTIGLIANO)Document20 pagesCalcul Des Déformations Par Les Méthodes Énergétiques (CASTIGLIANO)MsdPas encore d'évaluation
- Tpcourscc2 Cor 0910Document2 pagesTpcourscc2 Cor 0910Nacir DaikhPas encore d'évaluation
- Chapitre IIDocument4 pagesChapitre IIdihia didaPas encore d'évaluation
- Présentation Deuxiéme PrincipeDocument57 pagesPrésentation Deuxiéme Principered maxPas encore d'évaluation
- Chimie CoursDocument40 pagesChimie CoursJoachim BidallierPas encore d'évaluation
- Formulaire de Physique ExtraitDocument17 pagesFormulaire de Physique Extraitousseynou dialloPas encore d'évaluation
- Série 3 CorrigéDocument16 pagesSérie 3 Corrigéمحمد الأمين ولد عالي ابليلPas encore d'évaluation
- Res Thermo 2 2008Document2 pagesRes Thermo 2 2008hadjirakermetPas encore d'évaluation
- Formule ThermoélasticitéDocument8 pagesFormule ThermoélasticitéRifi Mohamed100% (1)
- MDF - TD1 CorrigéDocument5 pagesMDF - TD1 CorrigéBenjamin Édouard YargaPas encore d'évaluation
- Cours PHY 302Document12 pagesCours PHY 302Charles AyikutuPas encore d'évaluation
- Resume Themo1Document8 pagesResume Themo1suumiPas encore d'évaluation
- 2ème 3ème Principes ThermoDocument9 pages2ème 3ème Principes ThermoIslem DhifallahPas encore d'évaluation
- ExerciceDocument2 pagesExercicegc.ensam.2023Pas encore d'évaluation
- Thermodynamique ChimiqueDocument31 pagesThermodynamique Chimiquebanzanzadrelvan4Pas encore d'évaluation
- O1.2 Exp2 - Thermodynamique Part 2Document73 pagesO1.2 Exp2 - Thermodynamique Part 2Lise Maelle NGAMALEU NOUBISSIEPas encore d'évaluation
- Modélisation Mathématique CoursDocument17 pagesModélisation Mathématique CoursSaif Eddine Majdoub100% (1)
- RegimeVariable Partie1Document18 pagesRegimeVariable Partie1Sk CissePas encore d'évaluation
- Principes de ThermodynamiqueDocument15 pagesPrincipes de Thermodynamiquenajim68100% (1)
- Points Cours_Mouvement Dans Un Champ UniformeDocument6 pagesPoints Cours_Mouvement Dans Un Champ UniformesocranedeoufPas encore d'évaluation
- Genie EnvironnementDocument4 pagesGenie EnvironnementBasmã AlilechePas encore d'évaluation
- Microbiologie Environnementale Partie 4Document46 pagesMicrobiologie Environnementale Partie 4Basmã AlilechePas encore d'évaluation
- Master II Traitement Des EauxDocument3 pagesMaster II Traitement Des EauxBasmã AlilechePas encore d'évaluation
- Correction TD 2Document6 pagesCorrection TD 2Linda KoundziPas encore d'évaluation
- Binaire PiDocument9 pagesBinaire PiBasmã Alileche100% (1)
- Correction TDDocument5 pagesCorrection TDBasmã AlilechePas encore d'évaluation
- Chapitre VII AdoucissementDocument51 pagesChapitre VII AdoucissementBasmã AlilechePas encore d'évaluation
- IntroductionDocument3 pagesIntroductionBasmã AlilechePas encore d'évaluation
- Chap 1 Écologie AppliDocument18 pagesChap 1 Écologie AppliBasmã AlilechePas encore d'évaluation
- Analyse de Cycle de Vie (Acv) Chapitre 1-1Document53 pagesAnalyse de Cycle de Vie (Acv) Chapitre 1-1Basmã AlilechePas encore d'évaluation
- Version Final Polymere PDFDocument65 pagesVersion Final Polymere PDFyahya akkaouiPas encore d'évaluation
- Titrage VinaigreDocument2 pagesTitrage VinaigreDjahid Jo100% (2)
- TP Orga2Document6 pagesTP Orga2chaimaa12Pas encore d'évaluation
- Conclusiones de PlomoDocument14 pagesConclusiones de Plomomaria alejandra diaz peñuela0% (1)
- Rapport Stage - L'Étude de L'unité de Lavage Et L'unité de Flottation.Document49 pagesRapport Stage - L'Étude de L'unité de Lavage Et L'unité de Flottation.Stage OCP57% (7)
- DRA 08 - Rapport Oméga Explosion Confinée de Gaz v1 - 0Document121 pagesDRA 08 - Rapport Oméga Explosion Confinée de Gaz v1 - 0Share PointPas encore d'évaluation
- 210307-Expertise InfiltrationsDocument7 pages210307-Expertise Infiltrationsmehdiben86Pas encore d'évaluation
- 3.propriétés Et TechniquesDocument29 pages3.propriétés Et Techniquessoumia lefkirPas encore d'évaluation
- Effet Des Rayonnements IonisantsDocument34 pagesEffet Des Rayonnements Ionisantsjavi_de_garciaPas encore d'évaluation
- GLOBO Vision7 423-550Document128 pagesGLOBO Vision7 423-550acportfolioPas encore d'évaluation
- Cetiat Catalogue Formation Internet0 PDFDocument101 pagesCetiat Catalogue Formation Internet0 PDFfifi foufouPas encore d'évaluation
- TD N°2 Chimie 01Document4 pagesTD N°2 Chimie 01All AhmeDciaPas encore d'évaluation
- Le Miracle Du FerDocument4 pagesLe Miracle Du FerbainkssPas encore d'évaluation
- 04 Alchol, Phenol and Ether Set Test Final EDocument3 pages04 Alchol, Phenol and Ether Set Test Final Eummer farooqPas encore d'évaluation
- TDN°1Document2 pagesTDN°1عمر أوصيفPas encore d'évaluation
- FJ Fiche Bague de Guidage Beca 006 Piston 098051200 1713 11052015Document4 pagesFJ Fiche Bague de Guidage Beca 006 Piston 098051200 1713 11052015DarkedgePas encore d'évaluation
- Elasticité Chapitre 6Document21 pagesElasticité Chapitre 6Anis LepicPas encore d'évaluation
- Memoire Mud LoggingDocument82 pagesMemoire Mud LoggingLokman DridahPas encore d'évaluation
- Guide Subjectif de La 1ère Année de Médecine - Hadjer SebihiDocument8 pagesGuide Subjectif de La 1ère Année de Médecine - Hadjer SebihiWissal Elbar100% (4)
- Note de Calcul Assemblage Type 4Document8 pagesNote de Calcul Assemblage Type 4Abdelilah ElmahsaniPas encore d'évaluation
- Organisation Des Enseignements Filière: SMA (S3) Nature de L'enseignement: COURS Et TDDocument9 pagesOrganisation Des Enseignements Filière: SMA (S3) Nature de L'enseignement: COURS Et TDAssoumatiAzeddinePas encore d'évaluation
- G. Lamarre SimonDocument205 pagesG. Lamarre SimonNoussaPas encore d'évaluation
- Cours 5 C TRAFI 2BACDocument18 pagesCours 5 C TRAFI 2BACEnidroun OutPas encore d'évaluation
- Technique Du VideDocument90 pagesTechnique Du VideAbdelmajid Elmansouri100% (1)
- Capture D'écran . 2023-01-04 À 01.10.15Document7 pagesCapture D'écran . 2023-01-04 À 01.10.15امازيغي حرPas encore d'évaluation
- Chimie ActivitesDocument12 pagesChimie ActivitesEl Youbi MohammedPas encore d'évaluation
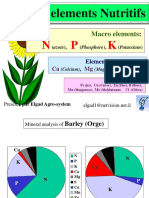
- Role Des Elements NutritifsDocument97 pagesRole Des Elements NutritifsFatre 1980Pas encore d'évaluation
- 3 EmeDocument1 page3 EmestefanPas encore d'évaluation
- Iso 1841 2 1996 PDFDocument8 pagesIso 1841 2 1996 PDFoubaha happyPas encore d'évaluation
- Rapport de Visite ShituruDocument4 pagesRapport de Visite ShituruDieu-Merci Kafutshi TshiyukaPas encore d'évaluation
- La vie des abeilles: Prix Nobel de littératureD'EverandLa vie des abeilles: Prix Nobel de littératureÉvaluation : 4 sur 5 étoiles4/5 (41)
- Améliorer votre mémoire: Un Guide pour l'augmentation de la puissance du cerveau, utilisant des techniques et méthodesD'EverandAméliorer votre mémoire: Un Guide pour l'augmentation de la puissance du cerveau, utilisant des techniques et méthodesÉvaluation : 5 sur 5 étoiles5/5 (2)
- L'Art de la guerre: Traité de stratégie en 13 chapitres (texte intégral)D'EverandL'Art de la guerre: Traité de stratégie en 13 chapitres (texte intégral)Évaluation : 4 sur 5 étoiles4/5 (3032)
- Secrets ancestraux d'un maître guérisseur: Un sceptique occidental, un maître oriental et les plus grands secrets de la vieD'EverandSecrets ancestraux d'un maître guérisseur: Un sceptique occidental, un maître oriental et les plus grands secrets de la vieÉvaluation : 5 sur 5 étoiles5/5 (2)
- Le profilage au service du football: Démarche scientifique pour un recrutement et entraînements optimisésD'EverandLe profilage au service du football: Démarche scientifique pour un recrutement et entraînements optimisésPas encore d'évaluation
- Électrotechnique | Pas à Pas: Bases, composants & circuits expliqués pour les débutantsD'EverandÉlectrotechnique | Pas à Pas: Bases, composants & circuits expliqués pour les débutantsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Semer avec succès pour rassembler avec abundance. Jardin organique et synergique: Calcul des meilleurs jours pour l'ensemencement de chaque légumeD'EverandSemer avec succès pour rassembler avec abundance. Jardin organique et synergique: Calcul des meilleurs jours pour l'ensemencement de chaque légumePas encore d'évaluation
- Harmonisation Energétique des Personnes: Manuel de Curothérapie 2020D'EverandHarmonisation Energétique des Personnes: Manuel de Curothérapie 2020Évaluation : 4 sur 5 étoiles4/5 (8)
- Un régime quantiqueD'EverandUn régime quantiqueÉvaluation : 5 sur 5 étoiles5/5 (1)
- 20 Véritables remèdes de nos grands-mères pour maigrir vite et enfin perdre du poidsD'Everand20 Véritables remèdes de nos grands-mères pour maigrir vite et enfin perdre du poidsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Transformez votre vie: Utilisez le pouvoir créateur qui est en vous pour construire votre vie à l'image de ce que vous voulez qu'elle soitD'EverandTransformez votre vie: Utilisez le pouvoir créateur qui est en vous pour construire votre vie à l'image de ce que vous voulez qu'elle soitÉvaluation : 4 sur 5 étoiles4/5 (14)
- Manuel de fabrication du savon: Je fabrique mes savons facilementD'EverandManuel de fabrication du savon: Je fabrique mes savons facilementÉvaluation : 5 sur 5 étoiles5/5 (4)
- Technologie automobile: Les Grands Articles d'UniversalisD'EverandTechnologie automobile: Les Grands Articles d'UniversalisPas encore d'évaluation
- L'Ombre à l'Univers: La structure des particules élémentaires XIIfD'EverandL'Ombre à l'Univers: La structure des particules élémentaires XIIfPas encore d'évaluation
- Approvisionnement et traitement de l’eau: Les Grands Articles d'UniversalisD'EverandApprovisionnement et traitement de l’eau: Les Grands Articles d'UniversalisPas encore d'évaluation
- Anatomie & 100 étirements essentiels pour le running: Principes de base, Techniques, Tableaux de séries, Précautions à prendre, Conseils, Programmes d'étirementsD'EverandAnatomie & 100 étirements essentiels pour le running: Principes de base, Techniques, Tableaux de séries, Précautions à prendre, Conseils, Programmes d'étirementsPas encore d'évaluation
- Géologie de l'Amérique: Les Grands Articles d'UniversalisD'EverandGéologie de l'Amérique: Les Grands Articles d'UniversalisPas encore d'évaluation
- L'Art de La Magie au Bougie Wicca: Le Guide du Débutant à la Pratique de la Magie au Bougie de WiccaD'EverandL'Art de La Magie au Bougie Wicca: Le Guide du Débutant à la Pratique de la Magie au Bougie de WiccaÉvaluation : 3 sur 5 étoiles3/5 (1)
- 160 ressources pour se lancer dans la vidéo quand on n’y connait rienD'Everand160 ressources pour se lancer dans la vidéo quand on n’y connait rienPas encore d'évaluation
- 500 secrets pour avoir un potager merveilleuxD'Everand500 secrets pour avoir un potager merveilleuxÉvaluation : 2 sur 5 étoiles2/5 (1)
- Harmonisation Energétique des Lieux: Habitat et haut-lieux sacrés 2020D'EverandHarmonisation Energétique des Lieux: Habitat et haut-lieux sacrés 2020Évaluation : 2.5 sur 5 étoiles2.5/5 (3)
- Jus de Fruits et de Légumes Crus: 57 recettes faciles et un Guide Pratique Complet pour améliorer votre alimentation .: Santé, Vitalité et Minceur, avec ... ET DURABLEMENTD'EverandJus de Fruits et de Légumes Crus: 57 recettes faciles et un Guide Pratique Complet pour améliorer votre alimentation .: Santé, Vitalité et Minceur, avec ... ET DURABLEMENTPas encore d'évaluation