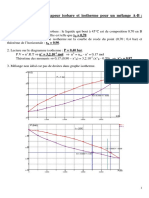
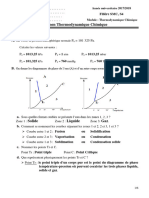
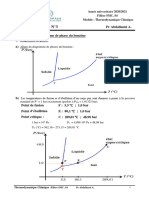
Vous aimerez peut-être aussi
- Détermination de La Masse Molaire Par CryoscopieDocument5 pagesDétermination de La Masse Molaire Par Cryoscopieeve100% (1)
- Diagrammes D EquilibreDocument36 pagesDiagrammes D EquilibreFrodon SacquetPas encore d'évaluation
- Bouhaouss-S4-Cha.i - II Et III Et IV 2015-16Document58 pagesBouhaouss-S4-Cha.i - II Et III Et IV 2015-16Rihab khPas encore d'évaluation
- Liquefaction D'un Gaz PDFDocument15 pagesLiquefaction D'un Gaz PDFKHALEDFEKAIRPas encore d'évaluation
- Cours Diagrammes Binaires Liquide-Vapeur-1Document71 pagesCours Diagrammes Binaires Liquide-Vapeur-1MOHAMED MAZOUARIPas encore d'évaluation
- Poly 4 WebDocument19 pagesPoly 4 Webbidounou essianePas encore d'évaluation
- Chap 3Document32 pagesChap 3souahila guesmiPas encore d'évaluation
- Chap2 Corps-PurDocument23 pagesChap2 Corps-Purait hssainPas encore d'évaluation
- CH I Equilibre Liquide VapeurDocument10 pagesCH I Equilibre Liquide Vapeurlo100% (1)
- 2012 Cours 4Document32 pages2012 Cours 4Roi AroufPas encore d'évaluation
- Cours - 3 Quilibres-SolubilitDocument13 pagesCours - 3 Quilibres-Solubilitamina mezouaghPas encore d'évaluation
- TH6 MachinesThermiques MPSIDocument7 pagesTH6 MachinesThermiques MPSIYouness HassibPas encore d'évaluation
- 2e Principe de La ThermoDocument15 pages2e Principe de La ThermoMohamed MohPas encore d'évaluation
- Ch21 CP StudentsDocument23 pagesCh21 CP StudentsSoulaima MatmatiPas encore d'évaluation
- Changement D Etat Du Corps Pur CoursDocument4 pagesChangement D Etat Du Corps Pur Courspicasso da vinciPas encore d'évaluation
- Chapitre 5 Diagramme de Phases de Corps Purs-1Document10 pagesChapitre 5 Diagramme de Phases de Corps Purs-1Rayen Jlassi100% (1)
- Corps PurDocument5 pagesCorps Purtraadel_320610041Pas encore d'évaluation
- Thermo Etude Thermo VapeursDocument7 pagesThermo Etude Thermo VapeursJijou HamidiPas encore d'évaluation
- Cours Thermodynamique - Chap 2Document36 pagesCours Thermodynamique - Chap 2Lavd LoghPas encore d'évaluation
- Thermodyanamique Appliquée Chapitre 3Document10 pagesThermodyanamique Appliquée Chapitre 3Rafik BourenanePas encore d'évaluation
- Équilibre de Phases Des Corps PursDocument17 pagesÉquilibre de Phases Des Corps PursSTOLEN MEMESPas encore d'évaluation
- Chapitre 4-1 Corps Pur EtudiantcorrigeDocument3 pagesChapitre 4-1 Corps Pur EtudiantcorrigeAriel AdepoPas encore d'évaluation
- Chap II Aspect Cinétique de La ThermodynamiqueDocument21 pagesChap II Aspect Cinétique de La ThermodynamiqueJean Marc LengePas encore d'évaluation
- Thermo Diagrammes Thermo PDFDocument7 pagesThermo Diagrammes Thermo PDFزكرياء بنحيرتPas encore d'évaluation
- Th. Cl. Chapitre XDocument13 pagesTh. Cl. Chapitre XAissiou NabilaPas encore d'évaluation
- Document Sans TitreDocument7 pagesDocument Sans TitreMohamed MahdiPas encore d'évaluation
- 8 - Equilibre liquide - vapeurDocument52 pages8 - Equilibre liquide - vapeurEtienne jeoffreyPas encore d'évaluation
- Changement D'étatDocument7 pagesChangement D'étatAhmed Yassine BouchanaPas encore d'évaluation
- Chapitre 12Document4 pagesChapitre 12trikiPas encore d'évaluation
- Thermo c8 SiteDocument28 pagesThermo c8 SiteAmin LarouiPas encore d'évaluation
- Pres ChapI Gaz RéelsDocument29 pagesPres ChapI Gaz RéelsHamadou Seidou MounkaÏla GoudelPas encore d'évaluation
- Chapitre IIIDocument16 pagesChapitre IIIAPas encore d'évaluation
- ST2 Thermodynamique GourariDocument38 pagesST2 Thermodynamique GouraritthPas encore d'évaluation
- Chapitre 1Document25 pagesChapitre 1Nourelhouda SELMANEPas encore d'évaluation
- Diagramme BinaireDocument20 pagesDiagramme BinaireجعدبندرهمPas encore d'évaluation
- Cours Réactivité Chimique BCG S2 (2020)Document198 pagesCours Réactivité Chimique BCG S2 (2020)Casa AbdessamadPas encore d'évaluation
- L 314Document23 pagesL 314Abderrahmen HassounaPas encore d'évaluation
- Résumé ThermochimieDocument9 pagesRésumé ThermochimiejeanPas encore d'évaluation
- Cours Résumé ThermodyDocument18 pagesCours Résumé Thermodyhabib benahmedPas encore d'évaluation
- Chap 2 Thermodynamique GAZ REELSDocument39 pagesChap 2 Thermodynamique GAZ REELSNour El Houda TebbanePas encore d'évaluation
- Chapitre 4-Transitions de Phases Dun Corps PurDocument24 pagesChapitre 4-Transitions de Phases Dun Corps PurbfbrfvlfrerPas encore d'évaluation
- Cours Procédés de Dépollution S6Document40 pagesCours Procédés de Dépollution S6CHAIMAEPas encore d'évaluation
- Diagramme de PhasesDocument98 pagesDiagramme de Phaseskabli ilyassPas encore d'évaluation
- 3-Le Second Principe de La ThermodynamiqueDocument12 pages3-Le Second Principe de La ThermodynamiqueAntes de Partir, A.C.Pas encore d'évaluation
- CH 2 - ThermoDocument14 pagesCH 2 - ThermoHaniPas encore d'évaluation
- Chapitre III SéchageDocument6 pagesChapitre III SéchageayadiPas encore d'évaluation
- Chapitre 1Document5 pagesChapitre 1wasimPas encore d'évaluation
- 8 Thermodynamique - Chimique Resume 3Document11 pages8 Thermodynamique - Chimique Resume 3Sell LarbiPas encore d'évaluation
- Formule de ClapeyronDocument7 pagesFormule de ClapeyronakaabadrePas encore d'évaluation
- C4cvl ENSTADocument17 pagesC4cvl ENSTAbensouiciPas encore d'évaluation
- Thermodynamique1 PDFDocument19 pagesThermodynamique1 PDFabdelmoumen.doudoPas encore d'évaluation
- Cours de Distillation DefDocument25 pagesCours de Distillation DefFars AminePas encore d'évaluation
- Equilibre Liquide - VapeurDocument12 pagesEquilibre Liquide - VapeurOussama ChahirPas encore d'évaluation
- Statique Des Fluides-CorrDocument7 pagesStatique Des Fluides-CorrCharaf LeoPas encore d'évaluation
- Rappel TH Série 3Document6 pagesRappel TH Série 3Yacine RMPas encore d'évaluation
- CH 5Document14 pagesCH 5salma.souissiPas encore d'évaluation
- ThermodynamiqueDocument8 pagesThermodynamiqueTess Berthier100% (1)
- Cours 13 Équilibres ChimiquesDocument4 pagesCours 13 Équilibres ChimiquesArti80% (5)
- Cours Thermodynamique - Chap 3Document21 pagesCours Thermodynamique - Chap 3Lavd LoghPas encore d'évaluation
- Chapitre 3Document14 pagesChapitre 3Junias PakiPas encore d'évaluation
- Distillation Fractionnée D'un Mélange Binaire (Eau - Acétone)Document4 pagesDistillation Fractionnée D'un Mélange Binaire (Eau - Acétone)achhamoudaPas encore d'évaluation
- DiagrammeDocument5 pagesDiagrammeBesma HamdiPas encore d'évaluation
- 5 - Diagrammes de Phases Binaires - Métaux - SDMGMPDocument11 pages5 - Diagrammes de Phases Binaires - Métaux - SDMGMPDjm Alg100% (1)
- Diagrammes de Phases BISDocument108 pagesDiagrammes de Phases BISSophia AvaPas encore d'évaluation
- 1.8 Binaire Solide Liquide Cours InverséDocument11 pages1.8 Binaire Solide Liquide Cours InverséOumeyma HamlauiPas encore d'évaluation
- Chimie Descriptive I Diagrammes de Phases Test 02Document16 pagesChimie Descriptive I Diagrammes de Phases Test 02Au RevoirPas encore d'évaluation
- O2 - Binaires G2D CorrigeDocument8 pagesO2 - Binaires G2D Corrigeoumayma afdhalPas encore d'évaluation
- Examen S4 Mai 2018 - Solution DétailléeDocument6 pagesExamen S4 Mai 2018 - Solution DétailléeMalki imanePas encore d'évaluation
- Les Diagrammes Binaires TD Corrigé 2020-2021Document4 pagesLes Diagrammes Binaires TD Corrigé 2020-2021Hamza Boulika50% (2)
- Chapitre 4-3 Thermo EtudiantDocument2 pagesChapitre 4-3 Thermo EtudiantAriel AdepoPas encore d'évaluation
- Diagrammes - Solide - Liquide (Chimie)Document8 pagesDiagrammes - Solide - Liquide (Chimie)Chedly TrimechPas encore d'évaluation
- Le Corps Pur Et Ses Caracteristiques Cours 2Document3 pagesLe Corps Pur Et Ses Caracteristiques Cours 2elboussabiayman6Pas encore d'évaluation
- TD1-Complèmentaire-Corps PurDocument5 pagesTD1-Complèmentaire-Corps PurmennanesalaheddinePas encore d'évaluation
- Diagrammes Binaires Solide-Liquide PDFDocument3 pagesDiagrammes Binaires Solide-Liquide PDFAmine Hattal100% (2)
- Chapitre 10 Equilibre Liquide - VapeurDocument28 pagesChapitre 10 Equilibre Liquide - VapeurNd'a Evrard KonanPas encore d'évaluation
- Distillation de Mélanges Non Idéaux Distillation Azéotropique Et Distillation Extractive. Choix de L'entraîneurDocument28 pagesDistillation de Mélanges Non Idéaux Distillation Azéotropique Et Distillation Extractive. Choix de L'entraîneurdahmani inesPas encore d'évaluation
- Cours 2 - Le Corps Pur Et Ses CaractéristiquesDocument3 pagesCours 2 - Le Corps Pur Et Ses Caractéristiquesrlm551830Pas encore d'évaluation
- Série #3 - S4 2021 - SolutionDocument8 pagesSérie #3 - S4 2021 - Solutionharedwaiss2010Pas encore d'évaluation
- Cours Les Changements D'état Physique D'un Corps Pur - 1ere AnnéeDocument3 pagesCours Les Changements D'état Physique D'un Corps Pur - 1ere AnnéeSmaali Faouzi Smaali100% (5)
- Test 1 Diagramme de Phases SMC3Document17 pagesTest 1 Diagramme de Phases SMC3ali BourenanePas encore d'évaluation
- Equilibres 2Document16 pagesEquilibres 2Mohammed Elhabib ZellalPas encore d'évaluation
- Diagrammes de PhasesDocument35 pagesDiagrammes de PhasesEliz2604100% (3)
- Support de Cours de Diagramme de Phases SMC3Document65 pagesSupport de Cours de Diagramme de Phases SMC3Yc YacinePas encore d'évaluation
- Chapitre 3 OPU GPE DistillationDocument14 pagesChapitre 3 OPU GPE DistillationADJOUDJ AMELPas encore d'évaluation
- Test 1 Diagramme de Phases SMC3Document17 pagesTest 1 Diagramme de Phases SMC3oumayma afdhal100% (1)
- Diagramme Phase JMF Tern Partie 1 Etudiant 22 23Document67 pagesDiagramme Phase JMF Tern Partie 1 Etudiant 22 23Abdel AdagolodjoPas encore d'évaluation
- Equilibres Solide Liquide PDFDocument5 pagesEquilibres Solide Liquide PDFWael ZidPas encore d'évaluation
- Polycopier Cours Diagrammes PDFDocument13 pagesPolycopier Cours Diagrammes PDFKENZA AHERFOUCH100% (2)
- Sujet Examen d'Equilibre entre phases 2023-2024Document2 pagesSujet Examen d'Equilibre entre phases 2023-2024elongoPas encore d'évaluation