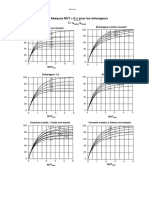
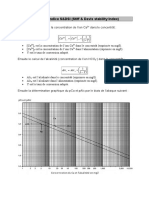
Vous aimerez peut-être aussi
- Cours Procédés de Dépollution S6Document40 pagesCours Procédés de Dépollution S6CHAIMAEPas encore d'évaluation
- Cours de Distillation DefDocument25 pagesCours de Distillation DefFars AminePas encore d'évaluation
- Cours Diagrammes Binaires Liquide-Vapeur-1Document71 pagesCours Diagrammes Binaires Liquide-Vapeur-1MOHAMED MAZOUARIPas encore d'évaluation
- TP4 Synthèse de L'acide Polylactique PLADocument1 pageTP4 Synthèse de L'acide Polylactique PLAahmeboudefirahmePas encore d'évaluation
- Note Chapitre 5 FluidisationDocument15 pagesNote Chapitre 5 FluidisationNawal AzrouPas encore d'évaluation
- Cours TransfrtDocument17 pagesCours TransfrtanisPas encore d'évaluation
- Manuel Extraction PulséeDocument29 pagesManuel Extraction PulséeMedPas encore d'évaluation
- UntitledDocument2 pagesUntitledRima mimiPas encore d'évaluation
- CalorimetrieDocument5 pagesCalorimetrieFaculté De Médecine BécharPas encore d'évaluation
- TD 3 de ThermochimieDocument2 pagesTD 3 de ThermochimieCARDI BPas encore d'évaluation
- Exposer de PétrochimieDocument37 pagesExposer de PétrochimieNoureddine FrhPas encore d'évaluation
- Clim1 DebitsDocument4 pagesClim1 DebitsYessine MrabetPas encore d'évaluation
- C1 Diagrammes Potentiel PH TD PDFDocument12 pagesC1 Diagrammes Potentiel PH TD PDFNourchaine MadiouniPas encore d'évaluation
- Polycopie TP TQM l3 GP 2021-2022Document3 pagesPolycopie TP TQM l3 GP 2021-2022Section E GP100% (1)
- Chapitre 1Document4 pagesChapitre 1Amina BadaouiPas encore d'évaluation
- Détermination de La Constante D'aciditéDocument3 pagesDétermination de La Constante D'aciditéAnis Dahnen100% (2)
- TP de Détermination de Masse Volumique Apparent Et AbsolueDocument6 pagesTP de Détermination de Masse Volumique Apparent Et AbsolueBen AbdePas encore d'évaluation
- Chimie de Sur2Document41 pagesChimie de Sur2Laiadhi DjemouiPas encore d'évaluation
- TD2 2018Document6 pagesTD2 2018Hassan ChehouaniPas encore d'évaluation
- Serie TDDocument4 pagesSerie TDTaoufik Ez100% (1)
- Chap 3 Diagrammes EpHDocument7 pagesChap 3 Diagrammes EpHayoub echraaPas encore d'évaluation
- Exercice GravitationDocument2 pagesExercice GravitationMadou TraorePas encore d'évaluation
- Mouhous OuahabDocument56 pagesMouhous OuahabMaamar LaidiPas encore d'évaluation
- Deux Pompes Identiques Placées en Série Puisent 2Document2 pagesDeux Pompes Identiques Placées en Série Puisent 2Fouad DimanePas encore d'évaluation
- Serie 2Document8 pagesSerie 2Mourad MatmourPas encore d'évaluation
- Phénomènes D'adsorptionDocument2 pagesPhénomènes D'adsorptionBouchraPas encore d'évaluation
- ERSA 04 (Ox) Al Sol AqueuseDocument4 pagesERSA 04 (Ox) Al Sol AqueuseAli AourdouPas encore d'évaluation
- TP Génie de La RéactionDocument4 pagesTP Génie de La RéactionImen ChibanePas encore d'évaluation
- Cours - MDF (Statique Cinématique) 22 23 (S1)Document27 pagesCours - MDF (Statique Cinématique) 22 23 (S1)Ghizlane100% (1)
- SM-BOUCHERDOUD - Ahmed-Chimie - Fondamentale-TP 3-Thermodynamique Et Cinétique Chimique - L2-S4Document2 pagesSM-BOUCHERDOUD - Ahmed-Chimie - Fondamentale-TP 3-Thermodynamique Et Cinétique Chimique - L2-S4fatimazahraPas encore d'évaluation
- Milieux Poreux Et Dispersés: 1. Le MateriauDocument5 pagesMilieux Poreux Et Dispersés: 1. Le MateriauAnesPas encore d'évaluation
- Traveaux Pratique OPU: Variation de La Perte de Charge Sur Une Colonne de Distillation DiscontinnueDocument9 pagesTraveaux Pratique OPU: Variation de La Perte de Charge Sur Une Colonne de Distillation Discontinnuenour nour100% (1)
- Binaires 2Document8 pagesBinaires 2محمد الفاتح100% (1)
- K Mansouri 2020Document57 pagesK Mansouri 2020Mourad MatmourPas encore d'évaluation
- Abaques NUT F (E)Document1 pageAbaques NUT F (E)aberranePas encore d'évaluation
- Dist Chap2Document12 pagesDist Chap2henryPas encore d'évaluation
- TPB13 CalorimetrieDocument3 pagesTPB13 CalorimetrieToufik Al HakimPas encore d'évaluation
- Chapitre IVet VDocument15 pagesChapitre IVet VÉtoile D'orPas encore d'évaluation
- UfasDocument7 pagesUfasRachid NaPas encore d'évaluation
- tp3 AdsDocument6 pagestp3 AdsMohamed EL AminePas encore d'évaluation
- Transformation de Laplace 2021Document14 pagesTransformation de Laplace 2021Msadak AnasPas encore d'évaluation
- TP MGC 131 N°2Document2 pagesTP MGC 131 N°2Ikram BenPas encore d'évaluation
- La Distillation - Partie 2-: Notes de Cours: Distillation - Mme BRAKCHIDocument7 pagesLa Distillation - Partie 2-: Notes de Cours: Distillation - Mme BRAKCHIHanane KhettabiPas encore d'évaluation
- TP Génie de ProcedésDocument22 pagesTP Génie de ProcedéskaltoumPas encore d'évaluation
- Série N°1 TD SDocument1 pageSérie N°1 TD SOussama BajaddaPas encore d'évaluation
- TP DE GENIE CHIMIQUE 2 (Absorption - Distillation) L3 GPDocument13 pagesTP DE GENIE CHIMIQUE 2 (Absorption - Distillation) L3 GPlamis b-zPas encore d'évaluation
- TP1 Génie PétrochimiqueDocument6 pagesTP1 Génie Pétrochimiqueياسمين لقرافPas encore d'évaluation
- Série 1Document2 pagesSérie 1slh 01Pas encore d'évaluation
- TP BernoulliDocument23 pagesTP BernoulliYasmine YasminePas encore d'évaluation
- TP FluDocument21 pagesTP FluHamza BejaouiPas encore d'évaluation
- M1 CP Examen 2021 SolutionDocument3 pagesM1 CP Examen 2021 Solutionsafia taibaouiPas encore d'évaluation
- Bibliographie DistllationDocument12 pagesBibliographie DistllationBrave ZinebPas encore d'évaluation
- La Structure Du SolDocument12 pagesLa Structure Du SolYassine BelkadaPas encore d'évaluation
- Cours 1 Bilans MacroscopiquesDocument3 pagesCours 1 Bilans MacroscopiquesLina alikhPas encore d'évaluation
- TP 1Document7 pagesTP 1Manar Youssef100% (1)
- TP 3 Théorème de Bernoulli CDocument5 pagesTP 3 Théorème de Bernoulli CMęlį SsæPas encore d'évaluation
- Polycope TP-TMDocument4 pagesPolycope TP-TMamiour krimo100% (1)
- Corrigé de TD2 - Pétrochimie 2 - L3 Raffinage Et PétrochimieDocument3 pagesCorrigé de TD2 - Pétrochimie 2 - L3 Raffinage Et Pétrochimieabdo myPas encore d'évaluation
- CM AtoL1 1 PDFDocument66 pagesCM AtoL1 1 PDFNicaise Amani YaoPas encore d'évaluation
- Principes de La Lyophilisation - Le LyophilisateurDocument250 pagesPrincipes de La Lyophilisation - Le Lyophilisateurjose sanchezPas encore d'évaluation
- TBA 2e 3 Chauffage Ventilation CompletDocument48 pagesTBA 2e 3 Chauffage Ventilation CompletSelin NartPas encore d'évaluation
- Modèle Weatherland DZABANA Phys 2Document22 pagesModèle Weatherland DZABANA Phys 2Presli KprtinoPas encore d'évaluation
- Cours Absorption +extraction Liquide Liquide 24-01-2021Document32 pagesCours Absorption +extraction Liquide Liquide 24-01-2021Fouad MihoubPas encore d'évaluation
- Règle Des Segments InversesDocument5 pagesRègle Des Segments InversesMouad GuendouzPas encore d'évaluation
- Exercices Corrigés en ChromatographieDocument13 pagesExercices Corrigés en ChromatographiePeti' Pou89% (66)
- SGE M1 Physico Chimie 2 3 2006Document51 pagesSGE M1 Physico Chimie 2 3 2006Mohamed CHIBANPas encore d'évaluation
- Tableau ChimieDocument38 pagesTableau Chimieemilie cyrPas encore d'évaluation
- Les PolymeresDocument9 pagesLes PolymeresfatihamPas encore d'évaluation
- Demande Inscription Oncours 2022Document4 pagesDemande Inscription Oncours 2022Ibrahima Mamadou DramePas encore d'évaluation
- TP3 - Thermo 2023Document12 pagesTP3 - Thermo 2023yeussefzerrad100% (1)
- Cofem Detector. 620002+A42NB - COFEM - C50SH-C50S-C50H-C50T - Instrucciones Uso - MUL - NOV21Document2 pagesCofem Detector. 620002+A42NB - COFEM - C50SH-C50S-C50H-C50T - Instrucciones Uso - MUL - NOV21corte001-omxPas encore d'évaluation
- Cinetique PDFDocument16 pagesCinetique PDFSamah SoltanePas encore d'évaluation
- IntroductionDocument5 pagesIntroductionDometanhan TuoPas encore d'évaluation
- 2-Série Acide Base + CorrigéDocument14 pages2-Série Acide Base + Corrigémimi mimiPas encore d'évaluation
- TP ChaleurDocument20 pagesTP ChaleurKaoutar CrusPas encore d'évaluation
- Orniformation Probatoire 1999 A PhysiqueDocument2 pagesOrniformation Probatoire 1999 A PhysiqueBoniface KouamPas encore d'évaluation
- BiochimieDocument3 pagesBiochimieb9rgpvqvmpPas encore d'évaluation
- CHP 01 - Les Semi-Conducteurs PDFDocument70 pagesCHP 01 - Les Semi-Conducteurs PDFDouaa BELHADJPas encore d'évaluation
- Dai Fichesureindustrie Reseauvapeur Edition2010Document22 pagesDai Fichesureindustrie Reseauvapeur Edition2010tazi kokoPas encore d'évaluation
- Procédure de CalculDocument17 pagesProcédure de CalculBaha-Eddine ElmoufadalPas encore d'évaluation
- Cours SDMDocument78 pagesCours SDMahmed ghassoulPas encore d'évaluation
- Unité de Réfrigeration IndustrielleDocument7 pagesUnité de Réfrigeration Industriellemarwan2nouichiPas encore d'évaluation
- TP Chimie PhysiqueDocument9 pagesTP Chimie PhysiquekhaledrevialPas encore d'évaluation
- Fonctionnement D Un Congelateur EnonceDocument5 pagesFonctionnement D Un Congelateur EnonceAbdePas encore d'évaluation
- 1 - ESIReims - Froid Dans Le Monde - 2020 - v2Document21 pages1 - ESIReims - Froid Dans Le Monde - 2020 - v2Ibrahim ZouinePas encore d'évaluation
- Vitesse de CorrosionDocument3 pagesVitesse de CorrosionAmirPas encore d'évaluation
- Révision Cristallo 01 - CorrigéDocument8 pagesRévision Cristallo 01 - Corrigéabdeladimelgouryani2023Pas encore d'évaluation
- EVALUATION N1 2023 PCT 3eme-1Document2 pagesEVALUATION N1 2023 PCT 3eme-1salahoud-dine soulayePas encore d'évaluation