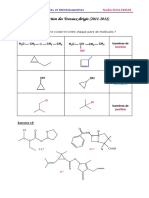
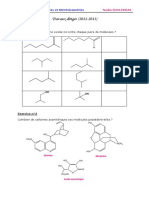
Vous aimerez peut-être aussi
- Acides Nucleiques Cours 02Document42 pagesAcides Nucleiques Cours 02MAITRE ValeryPas encore d'évaluation
- Antiépileptiques (Enregistrement Automatique)Document38 pagesAntiépileptiques (Enregistrement Automatique)nouha ninaPas encore d'évaluation
- Corrigé Série N°1 Exercices 4 Et 5Document4 pagesCorrigé Série N°1 Exercices 4 Et 5bouaddouanisPas encore d'évaluation
- Exercices-Chapitre-2 #BiolDocument5 pagesExercices-Chapitre-2 #BiolZinou Bourbia100% (1)
- Exercices Corrigés NomenclatureDocument4 pagesExercices Corrigés NomenclatureMalek abidi100% (5)
- Série-1 Corrigée Chimie4Document4 pagesSérie-1 Corrigée Chimie4poly educationPas encore d'évaluation
- 1 Les Antiépileptiques 2017Document56 pages1 Les Antiépileptiques 2017Na FesPas encore d'évaluation
- TDDocument10 pagesTDam47Pas encore d'évaluation
- Exercices IsomerieDocument10 pagesExercices IsomerieJean-François Abena100% (3)
- Acide CarboxderivesDocument40 pagesAcide CarboxderivesLevier PoidsPas encore d'évaluation
- SerieDocument3 pagesSeriechaima.sellamiPas encore d'évaluation
- PC C2013 CorDocument7 pagesPC C2013 Corrktm.avotraPas encore d'évaluation
- 22-23 Les Beta Lactamines Um6ss PR Benmoussa AdnaneDocument43 pages22-23 Les Beta Lactamines Um6ss PR Benmoussa AdnanesaloumaammorPas encore d'évaluation
- Glucide 1Document1 pageGlucide 1imane AGOURRAMPas encore d'évaluation
- Biologie Des Acides NucléiquesDocument22 pagesBiologie Des Acides Nucléiquesabekhti2008Pas encore d'évaluation
- Exercices NomenclatureDocument5 pagesExercices NomenclatureJean-François Abena71% (7)
- PHJDocument3 pagesPHJAbdullah MohamedPas encore d'évaluation
- TD Stereochimie Corrige 4Document3 pagesTD Stereochimie Corrige 4Aaouine AbderrazzakPas encore d'évaluation
- Séries TD-Chim Org-S1 (2020-2021)Document8 pagesSéries TD-Chim Org-S1 (2020-2021)Manel BahiPas encore d'évaluation
- LV203 Sujet Et Corrige 1er CC 2006Document4 pagesLV203 Sujet Et Corrige 1er CC 2006Za Hra100% (1)
- TD-chimie OrgaDocument21 pagesTD-chimie OrgaJean AnanPas encore d'évaluation
- Reactions D Esterification Et D Hydrolyse Cours LatexDocument5 pagesReactions D Esterification Et D Hydrolyse Cours LatexIsmail EloualiPas encore d'évaluation
- Chimie Organique TotalDocument69 pagesChimie Organique TotalCelestin KibishingoPas encore d'évaluation
- ++Exercices-Chapitre4 by ExoSupDocument9 pages++Exercices-Chapitre4 by ExoSupouiameabdelPas encore d'évaluation
- TD 2Document5 pagesTD 2Abdou HajjajiPas encore d'évaluation
- Exercice 1: Trouver Une Formule Semi-Développée Pour Chacun Des Composés SuivantsDocument2 pagesExercice 1: Trouver Une Formule Semi-Développée Pour Chacun Des Composés Suivantssteve dademPas encore d'évaluation
- EXAMEN 1 CORRIGéDocument3 pagesEXAMEN 1 CORRIGéSOUFYANE MUSTAPA100% (1)
- Exercices Corrigés NomenclatureDocument4 pagesExercices Corrigés Nomenclaturemedimegh.insaf2020Pas encore d'évaluation
- Chapitre 7 - Les Acides AminésDocument7 pagesChapitre 7 - Les Acides AminésBeatrice Florin100% (1)
- TD Biochimie FEVDocument52 pagesTD Biochimie FEVAbdou SaiPas encore d'évaluation
- Acide Alpha AminésDocument8 pagesAcide Alpha AminésMohamed BambaPas encore d'évaluation
- ANTIÉPILEPTIQUESDocument35 pagesANTIÉPILEPTIQUESTidjouguena Mawena CasimirPas encore d'évaluation
- Antituberculeux PDFDocument28 pagesAntituberculeux PDFMar YemPas encore d'évaluation
- Exercices AlcenesDocument9 pagesExercices AlcenesJean-François Abena33% (3)
- Exercices 41 CorrigeDocument11 pagesExercices 41 Corrige130660LYC OMAR BENABDELAZIZ NEDROMAPas encore d'évaluation
- TD NomenclatureDocument6 pagesTD NomenclatureSamba Barham100% (1)
- Video 53Document10 pagesVideo 53Dr VerdasPas encore d'évaluation
- Exo ISI 13 14 NomenclatureDocument2 pagesExo ISI 13 14 NomenclatureJoseph 27Pas encore d'évaluation
- Famille Et Nomenclature Des Molecules OrganiquesDocument5 pagesFamille Et Nomenclature Des Molecules OrganiquesUlysse HubertPas encore d'évaluation
- TD Stereochimie 5Document5 pagesTD Stereochimie 5Jean TellPas encore d'évaluation
- Chimie Organique 2014 DR DabiréDocument87 pagesChimie Organique 2014 DR DabiréGaétan YaméogoPas encore d'évaluation
- Examen de Formulacic3b3n Orgc3a1nica 1 PDFDocument3 pagesExamen de Formulacic3b3n Orgc3a1nica 1 PDFPatricia ColladoPas encore d'évaluation
- TD Dus - 1 PDFDocument28 pagesTD Dus - 1 PDFwacabamaPas encore d'évaluation
- EX1Document6 pagesEX1zgazga amirPas encore d'évaluation
- 19 Dec 2021-Cotrol Scene 1Document7 pages19 Dec 2021-Cotrol Scene 1Lê Nguyễn Quỳnh TrangPas encore d'évaluation
- I Modification Post TranscriptionnelleDocument25 pagesI Modification Post TranscriptionnelleFarida SirimaPas encore d'évaluation
- Les AntihistaminiquesDocument27 pagesLes AntihistaminiquesAmine OuriemchiPas encore d'évaluation
- Exercices Effets ElectronDocument9 pagesExercices Effets ElectronJean-François Abena100% (3)
- Session1 06Document3 pagesSession1 06Augustin WiniguePas encore d'évaluation
- Serie de TD ChimieDocument5 pagesSerie de TD Chimieraouf teach 04Pas encore d'évaluation
- Corr EXAMSESSI 1 - SMC - S3 2014-15Document4 pagesCorr EXAMSESSI 1 - SMC - S3 2014-15ismailelpro2021Pas encore d'évaluation
- Série 2 Isomérie 2020-2021Document3 pagesSérie 2 Isomérie 2020-2021Ayoub HmPas encore d'évaluation
- 5-Structure Des Acides Nucléiques 2019-2020 (Mode de Compatibilité)Document26 pages5-Structure Des Acides Nucléiques 2019-2020 (Mode de Compatibilité)AsmaaPas encore d'évaluation
- Aldéhydes CétonesDocument56 pagesAldéhydes CétonesRoi AroufPas encore d'évaluation
- UntitledDocument3 pagesUntitledAbdeldjalil MayachePas encore d'évaluation
- PeptidesDocument7 pagesPeptidesAchwak BelfadelPas encore d'évaluation
- Microbiologie Examen Corrigé 4Document4 pagesMicrobiologie Examen Corrigé 4SOKAMTE TEGANG Alphonse100% (1)
- 2) Mitose Et MéioseDocument52 pages2) Mitose Et MéioseNaruto & SasukePas encore d'évaluation
- Appareil de GolgiDocument6 pagesAppareil de Golgiidrissjulian9Pas encore d'évaluation
- 05 Noyau-InterphasiqueDocument10 pages05 Noyau-InterphasiqueTttttPas encore d'évaluation
- Chapitre 2Document13 pagesChapitre 2chiraz biotechPas encore d'évaluation
- TD1 MetabolismeDocument1 pageTD1 MetabolismeyvesPas encore d'évaluation
- Fiche TD01Document1 pageFiche TD01solo gamerPas encore d'évaluation
- Extrait Les Marqueurs Moleculaires en Genetique EtDocument20 pagesExtrait Les Marqueurs Moleculaires en Genetique EtMohammed El Machmoul100% (1)
- 2016 Koffi YvonneDocument215 pages2016 Koffi YvonneBadr GourmahPas encore d'évaluation
- Puce A Adn1Document2 pagesPuce A Adn1Elkhatibi Fatima-EzzahraPas encore d'évaluation
- 12 Anabolisme Des Acides Gras Et Des Triglycerides1Document7 pages12 Anabolisme Des Acides Gras Et Des Triglycerides1Moise NdiayePas encore d'évaluation
- Cours Immunologie - 2ème Sciences Biologiques Section 01 - OUABEDDocument19 pagesCours Immunologie - 2ème Sciences Biologiques Section 01 - OUABEDDjihadnoorPas encore d'évaluation
- BiocatalyseDocument23 pagesBiocatalysemohamed benlaraguePas encore d'évaluation
- College Protestant Comp. Tle D - 074611Document2 pagesCollege Protestant Comp. Tle D - 074611GeremyPas encore d'évaluation
- Cours Enzymes Pr.+ERRAFIY+NadiaDocument35 pagesCours Enzymes Pr.+ERRAFIY+NadiahpcnqrqdtmPas encore d'évaluation
- TD N 1 Biologie MoléculaireDocument6 pagesTD N 1 Biologie Moléculairefirdaoussaoud22Pas encore d'évaluation
- GénotoxicitéDocument5 pagesGénotoxicitéIkram HamachePas encore d'évaluation
- Acides Aminés Pour Aller Plus LoinDocument15 pagesAcides Aminés Pour Aller Plus LoinCooper WaynePas encore d'évaluation
- Gels D'agaroseDocument4 pagesGels D'agarosemolandPas encore d'évaluation
- 101à200 QCM FMA1 Bio CellDocument12 pages101à200 QCM FMA1 Bio CellFéz EyPas encore d'évaluation
- TP Cycle Cellulaire DocumentsDocument5 pagesTP Cycle Cellulaire Documentsjamal el azmyPas encore d'évaluation
- BIO111 ChapitreI Suite 01Document5 pagesBIO111 ChapitreI Suite 01Berthe Aristide Ngue Nkongo100% (1)
- Vocabulaire ScientifiqueDocument2 pagesVocabulaire ScientifiquenshoumoubenyPas encore d'évaluation
- Generalité Sur ADNDocument4 pagesGeneralité Sur ADNMedecine DentairePas encore d'évaluation
- ExamDocument2 pagesExamRoukiyath AmoussaPas encore d'évaluation
- La Cellule PDFDocument4 pagesLa Cellule PDFnehemie nwayPas encore d'évaluation
- Physio1an-Interaction Ligand Recepteur2016Document18 pagesPhysio1an-Interaction Ligand Recepteur2016AmirMohamedYozmanePas encore d'évaluation
- INTRODUCTIONDocument7 pagesINTRODUCTIONmy computerPas encore d'évaluation
- Le Cycle CellulaireDocument30 pagesLe Cycle CellulaireMohamadou Lamine KanePas encore d'évaluation
- A Synthèse Des ProtéinesDocument3 pagesA Synthèse Des ProtéinesNadaPas encore d'évaluation