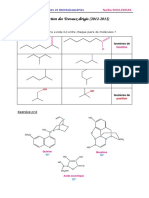
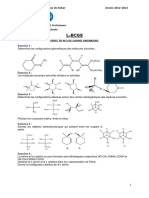
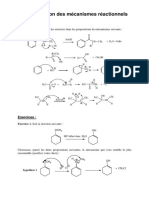
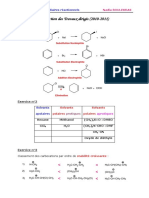
Vous aimerez peut-être aussi
- TD Infrarouge Corrigé 2020Document8 pagesTD Infrarouge Corrigé 2020Mohamed Belbaraka100% (6)
- CH Org TD 7Document8 pagesCH Org TD 7Mohammed El Kacem KadraouiPas encore d'évaluation
- TD Éffets Électroniques 2020Document3 pagesTD Éffets Électroniques 2020Berthe AmiPas encore d'évaluation
- Epreuve Juin smc6 14 15Document2 pagesEpreuve Juin smc6 14 15cvbn100% (1)
- Examen Correction L1 Chimie Organique 2006 2Document7 pagesExamen Correction L1 Chimie Organique 2006 2med madPas encore d'évaluation
- TD Stereochimie Corrige 5Document7 pagesTD Stereochimie Corrige 5othmanPas encore d'évaluation
- 2009 Lyon Walchshofer Radix StrucMolOrgaCours PDFDocument19 pages2009 Lyon Walchshofer Radix StrucMolOrgaCours PDFAbdessamed GherbaouiPas encore d'évaluation
- Pharmacologie EndodontiqueDocument9 pagesPharmacologie EndodontiqueKamalo TrissePas encore d'évaluation
- EXAMEN 1 CORRIGéDocument3 pagesEXAMEN 1 CORRIGéSOUFYANE MUSTAPA100% (2)
- Td11chim2019 2020Document3 pagesTd11chim2019 2020Med Stu DentPas encore d'évaluation
- Serie TD 2 L2BCGS 2022 - 2023-5Document2 pagesSerie TD 2 L2BCGS 2022 - 2023-5Daour NdiayePas encore d'évaluation
- Série 3 SMC4Document5 pagesSérie 3 SMC4anwar jakriPas encore d'évaluation
- Mecanismes Reactionnels PDFDocument6 pagesMecanismes Reactionnels PDFJérémie NimpagaritsePas encore d'évaluation
- Série 2 Isomérie 2020-2021Document3 pagesSérie 2 Isomérie 2020-2021Ayoub HmPas encore d'évaluation
- OrganometalliquesDocument13 pagesOrganometalliquesabdelouahed.rhaouiPas encore d'évaluation
- Corrige Session 2 2015 16Document7 pagesCorrige Session 2 2015 16Khalid ZegPas encore d'évaluation
- RevisionDocument10 pagesRevisionChimiste ChimistePas encore d'évaluation
- Session 1 2016Document7 pagesSession 1 2016Yc YacinePas encore d'évaluation
- 6 Reactions D Elimination 1Document9 pages6 Reactions D Elimination 1Blondet RomaricPas encore d'évaluation
- 15209cours Orga M1 CMS Reactions PericycliquesDocument33 pages15209cours Orga M1 CMS Reactions PericycliquesLaura DijouxPas encore d'évaluation
- Série-3-Corrigée Chimie4Document15 pagesSérie-3-Corrigée Chimie4poly educationPas encore d'évaluation
- 0 C 40 F 13 A 769 Aef 3 Ca 505Document4 pages0 C 40 F 13 A 769 Aef 3 Ca 505api-497754935Pas encore d'évaluation
- TD Nomenclature Corrigé12goodprepa PDFDocument3 pagesTD Nomenclature Corrigé12goodprepa PDFNour EddinePas encore d'évaluation
- Serie 3 PDFDocument2 pagesSerie 3 PDFElbahi Djaalab33% (3)
- SMC4 Examen Juin 2013Document5 pagesSMC4 Examen Juin 2013Yc YacinePas encore d'évaluation
- TD Intermediaires Reactionnels 2 SolutionDocument2 pagesTD Intermediaires Reactionnels 2 SolutionIssa SINDEPas encore d'évaluation
- Sujet Corrige Exam 2017Document6 pagesSujet Corrige Exam 2017huihu100% (1)
- 2016 Hétérocycles Caractères GénérauxDocument56 pages2016 Hétérocycles Caractères GénérauxhugoboddaertPas encore d'évaluation
- TDDocument10 pagesTDam47Pas encore d'évaluation
- Série 5Document1 pageSérie 5simo Gri100% (1)
- Nomenclature Exercices PDFDocument3 pagesNomenclature Exercices PDFmagnumPas encore d'évaluation
- S6 HOC CH Chapitre 4Document21 pagesS6 HOC CH Chapitre 4Eddouks Fatimazahrae100% (2)
- Chimie Organique Fonctionnelle TD 02Document2 pagesChimie Organique Fonctionnelle TD 02123456789Pas encore d'évaluation
- Al CoolsDocument13 pagesAl Coolsbmm2Pas encore d'évaluation
- A 3 CompetitionDocument3 pagesA 3 CompetitionEmilie BoubinetPas encore d'évaluation
- TD Orga3 L3 S5Document2 pagesTD Orga3 L3 S5DialloPas encore d'évaluation
- Chapitre 1 CHM321 CoordinationDocument35 pagesChapitre 1 CHM321 CoordinationVictor NguemoPas encore d'évaluation
- Chimie Organique Fonctionnelle TD Corr 01Document2 pagesChimie Organique Fonctionnelle TD Corr 01123456789Pas encore d'évaluation
- TD Chap 1-2Document8 pagesTD Chap 1-2Aris MpomePas encore d'évaluation
- Exam I 2Document1 pageExam I 2Anonymous sdeGjzXETG100% (2)
- TD Stereochimie 5Document5 pagesTD Stereochimie 5Jean TellPas encore d'évaluation
- TD1-composés Carbonylés IIIDocument3 pagesTD1-composés Carbonylés IIIRita No ExistePas encore d'évaluation
- Exam-Corrige Chimie PDFDocument4 pagesExam-Corrige Chimie PDFNassimaPas encore d'évaluation
- Exo TDDocument8 pagesExo TDBassirou YacoubaPas encore d'évaluation
- Exam I 7Document3 pagesExam I 7Dlimi MohamedPas encore d'évaluation
- Aldehydes CetonesDocument29 pagesAldehydes CetonesAhmad Elhamri100% (1)
- UntitledDocument3 pagesUntitledAbdeldjalil MayachePas encore d'évaluation
- Corrige Exercices 14 21 Chimie OrganiqueDocument33 pagesCorrige Exercices 14 21 Chimie OrganiqueFofo BokovouPas encore d'évaluation
- Aldéhydes Et CétonesDocument34 pagesAldéhydes Et Cétonesatika benslimanePas encore d'évaluation
- Serie1 Org2-1Document2 pagesSerie1 Org2-1Bibi Biba100% (1)
- DiolsDocument9 pagesDiolsAhmad ElhamriPas encore d'évaluation
- Diols PDFDocument9 pagesDiols PDFDaoui Ibtihel100% (2)
- Corrigé de Examen Final OrganiqueDocument2 pagesCorrigé de Examen Final OrganiqueRedOne DerrouazinPas encore d'évaluation
- Orga 6Document11 pagesOrga 6Romain Laher100% (1)
- Diagrammes Potentiel-PhDocument7 pagesDiagrammes Potentiel-Phaymane el hachimiPas encore d'évaluation
- TD Chim Orga FonctDocument11 pagesTD Chim Orga FonctAnäśś AnassPas encore d'évaluation
- TD SyntheseDocument11 pagesTD SyntheseibrahimaPas encore d'évaluation
- SMC4 Examen Juillet 2013Document4 pagesSMC4 Examen Juillet 2013Yc Yacine100% (1)
- TD Chapitre 8 SNDocument4 pagesTD Chapitre 8 SNزيد بخليفةPas encore d'évaluation
- Alcènes AlcynesDocument17 pagesAlcènes AlcynesINFINITY & BEYONDPas encore d'évaluation
- TD Substitution NucleophileDocument3 pagesTD Substitution NucleophileJohns AtsacwounPas encore d'évaluation
- Corrige Exercices 22 27 Chimie OrganiqueDocument20 pagesCorrige Exercices 22 27 Chimie Organiqueephraim MoukokoPas encore d'évaluation
- Universite D Abomey Calavi La Problematique de L Eau Potable Et La Sante Humaine Dans La Ville de Cotonou Odoulami Leocadie 2009 PDFDocument230 pagesUniversite D Abomey Calavi La Problematique de L Eau Potable Et La Sante Humaine Dans La Ville de Cotonou Odoulami Leocadie 2009 PDFزيد بخليفةPas encore d'évaluation
- La Problématique de L'eau en Algérie Enjeux Et Contraintes PDFDocument268 pagesLa Problématique de L'eau en Algérie Enjeux Et Contraintes PDFزيد بخليفة100% (1)
- DOERFLINGER 2017 Diffusion PDFDocument217 pagesDOERFLINGER 2017 Diffusion PDFزيد بخليفةPas encore d'évaluation
- Analyses Des Eaux de Réseau de La Ville de Béjaia Et Évaluation de Leur Pouvoir Entartant PDFDocument102 pagesAnalyses Des Eaux de Réseau de La Ville de Béjaia Et Évaluation de Leur Pouvoir Entartant PDFزيد بخليفة100% (1)
- Les MosommeDocument15 pagesLes Mosommeزيد بخليفةPas encore d'évaluation
- 12P14 PDFDocument156 pages12P14 PDFزيد بخليفةPas encore d'évaluation
- TD Chapitre 8 SNDocument4 pagesTD Chapitre 8 SNزيد بخليفةPas encore d'évaluation
- TD Substitution Nucleophile Corrige 3Document5 pagesTD Substitution Nucleophile Corrige 3زيد بخليفةPas encore d'évaluation
- Exercices HalogenoalcanesDocument10 pagesExercices HalogenoalcanesJean-François Abena100% (1)
- Rapport de StageDocument7 pagesRapport de Stageabdelghani benhamoudaPas encore d'évaluation
- TP MM TesDocument32 pagesTP MM TesAtayi100% (1)
- Compte Rendu de TPDocument4 pagesCompte Rendu de TPManonPas encore d'évaluation
- Devoir Chimie 1ere CDDocument3 pagesDevoir Chimie 1ere CDBriendon PogaPas encore d'évaluation
- Tse ChimieDocument67 pagesTse Chimiefombafousseini46Pas encore d'évaluation
- Travaux Dirigés de Chimie: Centre D'Encadrement ' Good Prepa''Document3 pagesTravaux Dirigés de Chimie: Centre D'Encadrement ' Good Prepa''Anta OndonPas encore d'évaluation
- Cours - Microbiologie Industrielle - BUT GB 2Document17 pagesCours - Microbiologie Industrielle - BUT GB 2Kérina FosPas encore d'évaluation
- Série D'exercices - Chimie Série Acide Base Acide Base - 2ème Sciences (2012-2013) MR Chouket HasenDocument2 pagesSérie D'exercices - Chimie Série Acide Base Acide Base - 2ème Sciences (2012-2013) MR Chouket HasenRamzi Tarchouni75% (4)
- 4 Propriétés Des OsesDocument20 pages4 Propriétés Des OsesMr. ZakariaPas encore d'évaluation
- 1ère A - APC - Les AlcanesDocument5 pages1ère A - APC - Les AlcanesLAWSON NICOLASPas encore d'évaluation
- Pfe AazmiDocument105 pagesPfe Aazmim'hamed elomari50% (2)
- Omposition de Himie NDocument15 pagesOmposition de Himie Namandine gaianiPas encore d'évaluation
- Essai de Multiplication Et Culture de Champignons Pleurote À Échelle Du LaboratoireDocument70 pagesEssai de Multiplication Et Culture de Champignons Pleurote À Échelle Du LaboratoireAMINEPas encore d'évaluation
- U 1000 RVFVDocument4 pagesU 1000 RVFVBouity MavyPas encore d'évaluation
- Cours Solubilite PDFDocument10 pagesCours Solubilite PDFRafik DraPas encore d'évaluation
- Objectifs Du Stage de Pharmacie HospitaliereDocument5 pagesObjectifs Du Stage de Pharmacie HospitaliereLolo ButyPas encore d'évaluation
- MINOUGOU Abdoul Kaled RachidDocument86 pagesMINOUGOU Abdoul Kaled RachiddanielsinkotPas encore d'évaluation
- Degradation PolymèreDocument5 pagesDegradation PolymèreHB RIMPas encore d'évaluation
- Guide de Nettoyage Et Entretien Du VerreDocument9 pagesGuide de Nettoyage Et Entretien Du VerreChristian NunezPas encore d'évaluation
- Écologie Bac 1 MédecineDocument38 pagesÉcologie Bac 1 Médecinemwambatshibanda04Pas encore d'évaluation
- Les Risques NRBC - Savoir Pour AgirDocument253 pagesLes Risques NRBC - Savoir Pour AgirazaPas encore d'évaluation
- Bio ImpoDocument20 pagesBio ImpoMeryem RkayaePas encore d'évaluation
- Vanne de DechargeDocument8 pagesVanne de DechargeSOFIANEPas encore d'évaluation
- Correction DS N°1 SpécialitéDocument2 pagesCorrection DS N°1 Spécialitédavid Bentouza100% (1)
- Huile BSE 85KDocument6 pagesHuile BSE 85KmithascoetPas encore d'évaluation
- 2012 Metropole Exo3 Correction CinetiqueConducti 4ptsDocument2 pages2012 Metropole Exo3 Correction CinetiqueConducti 4ptsĀbdï FïgõPas encore d'évaluation
- Exposé Sur:: Centre D'enfouissement Technique CET Des Déchets Urbains-Quel Avenir en AlgérieDocument20 pagesExposé Sur:: Centre D'enfouissement Technique CET Des Déchets Urbains-Quel Avenir en AlgériedrgocefPas encore d'évaluation
- GraphismeDocument5 pagesGraphismeabidi issam eddinePas encore d'évaluation