Vous aimerez peut-être aussi
- 1 3AM - phgenPrAlaoui21.22Document203 pages1 3AM - phgenPrAlaoui21.22Ismail BentaibiPas encore d'évaluation
- A - Developpement Des MedicamentsDocument63 pagesA - Developpement Des MedicamentsAmal Oulad AliPas encore d'évaluation
- Branches de La PharmacologieDocument8 pagesBranches de La PharmacologieradeyymvnprodPas encore d'évaluation
- Tox - Etudes Toxicologiques AlimentairesDocument6 pagesTox - Etudes Toxicologiques AlimentairesMed Khelloufi100% (4)
- Intro Vie Dun Médicament 2019Document56 pagesIntro Vie Dun Médicament 2019dhiadhayoutadhayouPas encore d'évaluation
- Cour 3Document106 pagesCour 3Abderrahmane DRISSIPas encore d'évaluation
- Cours de Toxicologie CHP6Document18 pagesCours de Toxicologie CHP6delégué microbiologiePas encore d'évaluation
- Chapitre 1Document24 pagesChapitre 1abdelmalekPas encore d'évaluation
- 1-Developpement Des MédicamentsDocument24 pages1-Developpement Des MédicamentsInés NamPas encore d'évaluation
- 1 - Introduction Médecine Dentaire 2019-2020Document13 pages1 - Introduction Médecine Dentaire 2019-2020See DownPas encore d'évaluation
- CM Medicaments-2022-PolyDocument13 pagesCM Medicaments-2022-PolysouennoughPas encore d'évaluation
- TOXICITEDocument6 pagesTOXICITESarah YahiaouiPas encore d'évaluation
- Cours Toxicologie AlimentaireDocument11 pagesCours Toxicologie AlimentaireJules RAKOTONARIVOPas encore d'évaluation
- Les Étapes de Développement Dun Nouveau MédicamentDocument4 pagesLes Étapes de Développement Dun Nouveau Médicamentfenrichahinez591Pas encore d'évaluation
- TOXICO Ticket GagnantDocument18 pagesTOXICO Ticket Gagnantchrys desoza elidadPas encore d'évaluation
- Notions PharmacologieDocument10 pagesNotions PharmacologieYoucef HoualefPas encore d'évaluation
- Généralités en Pharmacologie: F .Tardy - P .Audouy Promotion 2020/2023 - UE 2.11 Semestre 1Document70 pagesGénéralités en Pharmacologie: F .Tardy - P .Audouy Promotion 2020/2023 - UE 2.11 Semestre 1Mariah NakhassiPas encore d'évaluation
- Cours 1 Annee PharmacieDocument33 pagesCours 1 Annee PharmacieanaslayadPas encore d'évaluation
- Pharmacologie A3Document18 pagesPharmacologie A3A100% (1)
- Les Phases CliniquesDocument4 pagesLes Phases CliniquesMassouh Assoui100% (1)
- Am Cours de Pharmacologie 2018Document176 pagesAm Cours de Pharmacologie 2018wida abrPas encore d'évaluation
- Pharmacovigilance 3Document20 pagesPharmacovigilance 3pharmacologiegeneralePas encore d'évaluation
- Réponse Sérologique Du Chien Après PDFDocument6 pagesRéponse Sérologique Du Chien Après PDFMelina ayalaPas encore d'évaluation
- Enoncé Biologie Animale 2Document24 pagesEnoncé Biologie Animale 2Erdem pelicat GowanitPas encore d'évaluation
- Pharmacologie General1 4 PDFDocument32 pagesPharmacologie General1 4 PDFEmmanuelPas encore d'évaluation
- CT 3831 MenopurDocument9 pagesCT 3831 MenopurAllal EL AkhdarPas encore d'évaluation
- Chapitre 1 Notions Générales PharmacologieDocument25 pagesChapitre 1 Notions Générales PharmacologiekmeriemPas encore d'évaluation
- Ficheproduit 467 2023-04-15 123122Document4 pagesFicheproduit 467 2023-04-15 123122Talla MboupPas encore d'évaluation
- 06 - Processus de Recherche, Développement Et Production Du MédicamentDocument14 pages06 - Processus de Recherche, Développement Et Production Du Médicamenthicham1963Pas encore d'évaluation
- Chp1.2 - Développement Du Médicament (Enregistrement Automatique)Document52 pagesChp1.2 - Développement Du Médicament (Enregistrement Automatique)abdoulrazaksafrazPas encore d'évaluation
- Chp1.2 - Développement Du Médicament 24 01 19Document52 pagesChp1.2 - Développement Du Médicament 24 01 19z589ngrfq5Pas encore d'évaluation
- L'Essentiel en Pharmaco Gen (QE)Document10 pagesL'Essentiel en Pharmaco Gen (QE)Imad ImadPas encore d'évaluation
- TP SaraaDocument8 pagesTP SaraaYasmine YasminePas encore d'évaluation
- Pharmacocinetique QualitativeDocument45 pagesPharmacocinetique Qualitativecep100% (3)
- Recherche Sur Les Plantes - INSSA 2Document60 pagesRecherche Sur Les Plantes - INSSA 2Cynthia OuedraogoPas encore d'évaluation
- Pharmacotoxicologie - Chapitre I Introduction À La Pharmacologie PDFDocument7 pagesPharmacotoxicologie - Chapitre I Introduction À La Pharmacologie PDFM.B. IsmailPas encore d'évaluation
- Cours N°1Document15 pagesCours N°1relax relaxPas encore d'évaluation
- Investigation Toxicologique PDFDocument19 pagesInvestigation Toxicologique PDFLinaMimosaPas encore d'évaluation
- Pharmaco Toxicologie ExpérimentaleDocument45 pagesPharmaco Toxicologie ExpérimentaleMoncef CherifPas encore d'évaluation
- 2022s2l3bsa Pharmtox Data1Document32 pages2022s2l3bsa Pharmtox Data1Fella MazPas encore d'évaluation
- (UE6) LAROCHE - 2. Evaluation Des Médicaments CommercialisésDocument16 pages(UE6) LAROCHE - 2. Evaluation Des Médicaments CommercialisésPaul MarcelPas encore d'évaluation
- Toxicologie ExpérimentaleDocument57 pagesToxicologie ExpérimentaleOMAR EL HAMDAOUIPas encore d'évaluation
- TRANJ103 - TheorieDocument16 pagesTRANJ103 - Theoriemavrinissue4Pas encore d'évaluation
- N°1 - Le MédicamentDocument12 pagesN°1 - Le MédicamentemmamarquiegsmPas encore d'évaluation
- 1 Introduction G À La Pharmacologie S1Document6 pages1 Introduction G À La Pharmacologie S1Asmaa HrPas encore d'évaluation
- Evaluation de La ToxicitéDocument5 pagesEvaluation de La ToxicitéKhaled BouchaourPas encore d'évaluation
- 8.essais Cliniques (BB)Document5 pages8.essais Cliniques (BB)alae7004Pas encore d'évaluation
- Cours 3 Drug ScreeningDocument33 pagesCours 3 Drug ScreeningChawki MokademPas encore d'évaluation
- CM - ToxicologieDocument40 pagesCM - ToxicologiePaul Caro0% (1)
- Résumé Du Cours de La Biologie À La Médecine IIDocument40 pagesRésumé Du Cours de La Biologie À La Médecine IITeresa ZoorobPas encore d'évaluation
- Chap 5 PDFDocument9 pagesChap 5 PDFry yukPas encore d'évaluation
- 12 DMprocréationDocument9 pages12 DMprocréationDabé LaminePas encore d'évaluation
- PK IntroductionDocument40 pagesPK IntroductionOMAR EL HAMDAOUIPas encore d'évaluation
- Evaluation de La ToxiciteDocument11 pagesEvaluation de La ToxiciteSevinç KraliçesiPas encore d'évaluation
- Fiche Revision Pharmaco Ue 2.11 (2081)Document8 pagesFiche Revision Pharmaco Ue 2.11 (2081)Kelya Da cunhaPas encore d'évaluation
- Warda 2Document34 pagesWarda 2Assma EllPas encore d'évaluation
- Pharmacotoxicologie Guergour HDocument14 pagesPharmacotoxicologie Guergour HArchippe Abia TchangpinaPas encore d'évaluation
- Les Essais Cliniques 19 207092265608892463575 PDFDocument29 pagesLes Essais Cliniques 19 207092265608892463575 PDFPedro KalvinPas encore d'évaluation
- QE Pharmaco Version FinaleDocument13 pagesQE Pharmaco Version Finaletesnim chaouchPas encore d'évaluation
- L'infertilité N'est Pas Seulement Un Problème Du CoupleD'EverandL'infertilité N'est Pas Seulement Un Problème Du CouplePas encore d'évaluation
- Le Métabolisme Du CholestérolDocument21 pagesLe Métabolisme Du Cholestérolرزقي إبن فلسطينPas encore d'évaluation
- 21 Dermatoses Bulleuses - Cas CliniquesDocument208 pages21 Dermatoses Bulleuses - Cas CliniquesKada Ben youcefPas encore d'évaluation
- Les Quinze Premières Vies DHarry August (North Claire (North Claire) ) (Z-Library)Document404 pagesLes Quinze Premières Vies DHarry August (North Claire (North Claire) ) (Z-Library)Cécile AlexandriaPas encore d'évaluation
- 572 01088 PDFDocument79 pages572 01088 PDFmaroua henkaPas encore d'évaluation
- PE 13 14 PrepaDocument12 pagesPE 13 14 Prepamahmoud sfar hanchaPas encore d'évaluation
- Cours 5 PDFDocument47 pagesCours 5 PDFÑã DåPas encore d'évaluation
- 02 La Guilde Du CerisierDocument9 pages02 La Guilde Du CerisierHerbe De LunePas encore d'évaluation
- Alimentation Et État Nutritionnel Des Enfants de 0-36 Mois Dans La Commune Rurale de Sabotsy Namehana (MAHAZAFY - INSPC/2007)Document61 pagesAlimentation Et État Nutritionnel Des Enfants de 0-36 Mois Dans La Commune Rurale de Sabotsy Namehana (MAHAZAFY - INSPC/2007)HayZara MadagascarPas encore d'évaluation
- Cours de Biologie Les EnzymesDocument10 pagesCours de Biologie Les EnzymesOussamaPas encore d'évaluation
- Acide NucleiqueDocument4 pagesAcide NucleiqueAli GohPas encore d'évaluation
- Cours N1 Mecanisme D - Action Des Xenobiotiques 2016Document23 pagesCours N1 Mecanisme D - Action Des Xenobiotiques 2016souane ibrahim25% (4)
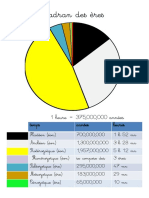
- Cadran Des ÈresDocument11 pagesCadran Des ÈresFrancofoliePas encore d'évaluation
- Le Palmier A HuileDocument6 pagesLe Palmier A HuileFrogy SebastianPas encore d'évaluation
- REVISION BIOS 202: Numéro 2 Exercice 1: Dans Un Récipient Contenant 1 Litre D'eau, On AjouteDocument2 pagesREVISION BIOS 202: Numéro 2 Exercice 1: Dans Un Récipient Contenant 1 Litre D'eau, On Ajouteyaa100% (1)
- Niveau X ClassificationsDocument4 pagesNiveau X ClassificationsMOZARGODWINPas encore d'évaluation
- Commentaire LectriceDocument3 pagesCommentaire LectriceSiham BouhaPas encore d'évaluation
- Cours de Biochimie Métabolisme Phosphocalcique Première Année Médecine DentaireDocument85 pagesCours de Biochimie Métabolisme Phosphocalcique Première Année Médecine DentaireNadar ArbiPas encore d'évaluation
- Klein 2014Document17 pagesKlein 2014ImanePas encore d'évaluation
- Filiere Pepiniere. de La Production A La Plantation. Innovations Techniques Produits MarchesDocument13 pagesFiliere Pepiniere. de La Production A La Plantation. Innovations Techniques Produits Marchesdjaouad bourouaisPas encore d'évaluation
- Devoir de Première S N°2 - Type II2Document3 pagesDevoir de Première S N°2 - Type II2Nihed BelhadjiPas encore d'évaluation
- Techniques D'immobilisation Des Cellules Et Leur ApplicationDocument50 pagesTechniques D'immobilisation Des Cellules Et Leur Applicationhouhou_houhou83% (24)
- (CCS) (2003) - ChimieDocument9 pages(CCS) (2003) - ChimiebabibenPas encore d'évaluation
- Les Plantes Utiles Du Gabon Journal D'agriculture Tropicale Etde Botanique AppliquéeDocument32 pagesLes Plantes Utiles Du Gabon Journal D'agriculture Tropicale Etde Botanique AppliquéekalyPas encore d'évaluation
- Evaluation Os MusclesDocument2 pagesEvaluation Os MusclesNiangPas encore d'évaluation
- Cardio HallaDocument53 pagesCardio HallaTewfik TGrPas encore d'évaluation
- Eckert - Fisiologia Animal PDFDocument621 pagesEckert - Fisiologia Animal PDFJesús LabGomPas encore d'évaluation
- Thyroide (Dr. Bouazdi 2016)Document68 pagesThyroide (Dr. Bouazdi 2016)Dounya BenyellesPas encore d'évaluation
- Null PDFDocument26 pagesNull PDFAziz OualidPas encore d'évaluation
- Capacité D'adaptation Au Changement Climatique À MadagascarDocument2 pagesCapacité D'adaptation Au Changement Climatique À MadagascarHayZara Madagascar100% (1)
- Chapitre 2 Tissu Conjonctif LAMDA 2021Document40 pagesChapitre 2 Tissu Conjonctif LAMDA 2021LàkàmoràEnAlgériePas encore d'évaluation