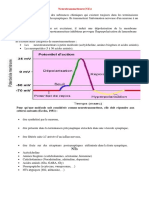
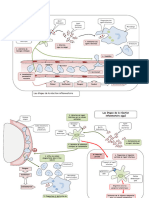
Vous aimerez peut-être aussi
- Neurobiologie: Les Grands Articles d'UniversalisD'EverandNeurobiologie: Les Grands Articles d'UniversalisPas encore d'évaluation
- Plan Definitive PDFDocument8 pagesPlan Definitive PDFManilaPas encore d'évaluation
- Herboristerie FamilialeDocument103 pagesHerboristerie Familialevilic93100% (1)
- Comment Détecter Les Poisons Mystiques Dans Les NourrituresDocument8 pagesComment Détecter Les Poisons Mystiques Dans Les Nourrituresaugustin NAGUIEBOUPas encore d'évaluation
- Le Taux Vibratoire PDFDocument15 pagesLe Taux Vibratoire PDFLuca TascaPas encore d'évaluation
- Les Champs MorphogenetiquesDocument12 pagesLes Champs MorphogenetiquesAnna Hamieux100% (2)
- Le Message Du DR Paul CartonDocument49 pagesLe Message Du DR Paul Cartonalvarez-duran.jesus6752Pas encore d'évaluation
- Facteurs Influençant Le Stress Oxydatif. Stress OxydatifDocument6 pagesFacteurs Influençant Le Stress Oxydatif. Stress OxydatifJean-Loïc BauchetPas encore d'évaluation
- La DesacidificationDocument50 pagesLa Desacidificationjuba1200100% (1)
- Le Grand Œuvre AlchimiqueDocument14 pagesLe Grand Œuvre Alchimiqueepoptae100% (1)
- La Transmission Synaptique 1Document6 pagesLa Transmission Synaptique 1safemindPas encore d'évaluation
- Transmission Synaptique PDFDocument8 pagesTransmission Synaptique PDFJean PatatePas encore d'évaluation
- QCM NeuroDocument15 pagesQCM NeuroJonathan JohnsonPas encore d'évaluation
- 2ème - physio.LA TRANSMISSION SYNAPTIQUE PDFDocument37 pages2ème - physio.LA TRANSMISSION SYNAPTIQUE PDFكوردورلي الحاجPas encore d'évaluation
- B.61-22 Reglage Des ProtectionsDocument37 pagesB.61-22 Reglage Des ProtectionsDazahra Med Nouh100% (1)
- Scion Supplément Panthéon ManittowockDocument26 pagesScion Supplément Panthéon ManittowockTerentius Avenelius PaladiusPas encore d'évaluation
- Définition, Concept, Approches en SantéDocument30 pagesDéfinition, Concept, Approches en Santésonia.bgh100% (1)
- Récepteurs MembranairesDocument56 pagesRécepteurs MembranairesAnonymous MKSfyYyODPPas encore d'évaluation
- Electro AcupunctureDocument13 pagesElectro AcupunctureCarlos MoreiraPas encore d'évaluation
- Devoir de Synthèse N°1 Collège Pilote - Français - 7ème (2008-2009) MR Gassoumi Mohamed LazharDocument5 pagesDevoir de Synthèse N°1 Collège Pilote - Français - 7ème (2008-2009) MR Gassoumi Mohamed LazharAhmed Massoudy50% (8)
- Cours Pharmacodynamie Etudiants2Document92 pagesCours Pharmacodynamie Etudiants2akizar Zinouk100% (1)
- Les Bouches Inutiles (Simone de Beauvoir)Document82 pagesLes Bouches Inutiles (Simone de Beauvoir)Omar MarínPas encore d'évaluation
- Réponses Exams BlancDocument5 pagesRéponses Exams BlancMourad Oujda100% (1)
- DevoirDocument9 pagesDevoirP EPas encore d'évaluation
- Cibles Moléculaires Et Élaboration de La Réponse 2024Document3 pagesCibles Moléculaires Et Élaboration de La Réponse 2024kaouaneamdjadPas encore d'évaluation
- La Liaison Aux RécepteursDocument8 pagesLa Liaison Aux RécepteursIsmaelPas encore d'évaluation
- Role Physiologique Des Neuromediateurs: DR Omar Dahmani, DR Amal Belcaid, DR Ouafa El Azzouzi, DR Hayat El HamiDocument5 pagesRole Physiologique Des Neuromediateurs: DR Omar Dahmani, DR Amal Belcaid, DR Ouafa El Azzouzi, DR Hayat El HamiathamosPas encore d'évaluation
- Chapitre 5signalisationDocument7 pagesChapitre 5signalisationFatïma ZohraPas encore d'évaluation
- Les Neurotransmetteurs PacesDocument30 pagesLes Neurotransmetteurs PacesValentin GeorgetPas encore d'évaluation
- 04 - La Transmission SynaptiqueDocument13 pages04 - La Transmission SynaptiqueNahla TrodiPas encore d'évaluation
- Resume de NeurophysiologieDocument5 pagesResume de NeurophysiologieMoussa DIAFOPas encore d'évaluation
- Synapse Et Transmission SynaptiqueDocument10 pagesSynapse Et Transmission SynaptiqueFaculté De Médecine BécharPas encore d'évaluation
- La Transmission SynaptiqueDocument39 pagesLa Transmission SynaptiquesmalldoctorivansPas encore d'évaluation
- 6 - Transmission SynaptiqueDocument7 pages6 - Transmission Synaptiquemintchimechak2Pas encore d'évaluation
- Pharmacodynamie, Cible, Mécanisme Daction Et Quantification de Leffet Du MédicamentDocument12 pagesPharmacodynamie, Cible, Mécanisme Daction Et Quantification de Leffet Du MédicamentPaul MarcelPas encore d'évaluation
- 6 Mécanisme D - Action Des Médicaments Médecine Dentaire 2022 2023Document34 pages6 Mécanisme D - Action Des Médicaments Médecine Dentaire 2022 2023makhlouf.farah02Pas encore d'évaluation
- Neuro 2Document12 pagesNeuro 2tinaPas encore d'évaluation
- Httpselearning Centre-Univ-Mila Dza2024pluginfile Php89198mod - resourcecontent1CHAPITRE20V PDFDocument20 pagesHttpselearning Centre-Univ-Mila Dza2024pluginfile Php89198mod - resourcecontent1CHAPITRE20V PDFariel.2mariemPas encore d'évaluation
- Chap II Récepteurs Membranaires Et Molécules de Signalisation IntracellulaireDocument28 pagesChap II Récepteurs Membranaires Et Molécules de Signalisation IntracellulaireAbdoulaye sebyPas encore d'évaluation
- 4-La Transmission Synaptique PDFDocument14 pages4-La Transmission Synaptique PDFNara ĪntiSsãrPas encore d'évaluation
- La Transmission Synaptique Medecine 2018Document10 pagesLa Transmission Synaptique Medecine 2018loulou liPas encore d'évaluation
- Question Type Examens Neurophysiologie PDFDocument10 pagesQuestion Type Examens Neurophysiologie PDFStephanePas encore d'évaluation
- 14 Histophysiologie Du Tissu NerveuxDocument7 pages14 Histophysiologie Du Tissu NerveuxGeorge BorsPas encore d'évaluation
- DCEM1 - Pharmacologie - Chapitre 19 - Les Antinicotiniques - Septembre 2005 PDFDocument4 pagesDCEM1 - Pharmacologie - Chapitre 19 - Les Antinicotiniques - Septembre 2005 PDFPaul fatheadPas encore d'évaluation
- Chapitre 1Document16 pagesChapitre 1David FMPas encore d'évaluation
- Cyto - Protéine G Et SignalisationDocument30 pagesCyto - Protéine G Et SignalisationOuarib ZakariaPas encore d'évaluation
- Mécanisme de La Transmission SynaptiqueDocument11 pagesMécanisme de La Transmission SynaptiqueAbubakr SalhiPas encore d'évaluation
- Cours 11 - La Cellule NerveuseDocument4 pagesCours 11 - La Cellule NerveuseSandra GomesPas encore d'évaluation
- MOSTA Physiologie de La Jonction NeuromusculaireDocument27 pagesMOSTA Physiologie de La Jonction NeuromusculaireSamia BelabbasPas encore d'évaluation
- Neurophysiologie Générale - Les Transmissions Synaptiques Lentes Et Les Cellules GlialesDocument10 pagesNeurophysiologie Générale - Les Transmissions Synaptiques Lentes Et Les Cellules GlialesanaischapellzPas encore d'évaluation
- Ligands Recepteur 2020 EtudiantDocument7 pagesLigands Recepteur 2020 EtudiantLina AmiraPas encore d'évaluation
- Interaction Ligand RecepteurDocument3 pagesInteraction Ligand RecepteurAmina MnPas encore d'évaluation
- Phrmacodynamie Suite DrDanDocument4 pagesPhrmacodynamie Suite DrDanDan NdiPas encore d'évaluation
- GP47Document1 pageGP47xgfhdu100% (1)
- Pharmacologie CanauxDocument2 pagesPharmacologie CanauxEl BakkaliPas encore d'évaluation
- 9-Système Renine Angiotensine AldostéroneDocument30 pages9-Système Renine Angiotensine AldostéroneRaïssa sawadogoPas encore d'évaluation
- TD Physiologique Endocrinienne. BABA & STAFFDocument13 pagesTD Physiologique Endocrinienne. BABA & STAFFmbouillesissoko55Pas encore d'évaluation
- Physio1an31-7jonction NeuromusculaireDocument15 pagesPhysio1an31-7jonction Neuromusculaireelidrissimaryam733Pas encore d'évaluation
- T. Nerveux 3. 1pptDocument50 pagesT. Nerveux 3. 1pptAdonis SerghiniPas encore d'évaluation
- GE 4 - CorrigéDocument6 pagesGE 4 - CorrigéalciaflsPas encore d'évaluation
- 08-Physiologie de La Jonction NeuromusculaireDocument26 pages08-Physiologie de La Jonction NeuromusculaireBook HunterPas encore d'évaluation
- PHARMACOLOGIE Des SystèmesDocument96 pagesPHARMACOLOGIE Des SystèmesRidou anePas encore d'évaluation
- Interaction Ligand RécepteurDocument6 pagesInteraction Ligand RécepteurFaculté De Médecine BécharPas encore d'évaluation
- Transduction Du Signal Et Régulation de La Fonction CellulaireDocument63 pagesTransduction Du Signal Et Régulation de La Fonction CellulaireMamadou FayePas encore d'évaluation
- Hormones CathecholaminesDocument8 pagesHormones Cathecholamineshananehadef04Pas encore d'évaluation
- Résumé GCDocument4 pagesRésumé GCLilia TlbPas encore d'évaluation
- Pompes, Transporteurs IoniqueDocument21 pagesPompes, Transporteurs IoniquedaoudPas encore d'évaluation
- 01 - La Communication IntercellulaireDocument8 pages01 - La Communication IntercellulaireHadil BenslamaPas encore d'évaluation
- Introduction Pharmaco. Generale m1 s1 G.PDocument13 pagesIntroduction Pharmaco. Generale m1 s1 G.PHamdaoui dounia100% (1)
- La Cellule Nerveuse W.PDF KKDocument6 pagesLa Cellule Nerveuse W.PDF KKas.cherfaouiPas encore d'évaluation
- RegulationDocument30 pagesRegulationYannick DsprbsPas encore d'évaluation
- Planches ElectrophysiologieDocument11 pagesPlanches Electrophysiologieleila chebahPas encore d'évaluation
- Tuyaux Neurophysio Àpd Anciens ExamsDocument19 pagesTuyaux Neurophysio Àpd Anciens ExamsjeannendangangPas encore d'évaluation
- Physio1an-Interactions Ligand Recepteur2021bougrida BoussoufDocument24 pagesPhysio1an-Interactions Ligand Recepteur2021bougrida BoussoufGI 2 Session 2018-2019Pas encore d'évaluation
- Le Dernier Defi de La Physique Moderne PDocument37 pagesLe Dernier Defi de La Physique Moderne PJean-Loïc BauchetPas encore d'évaluation
- Vortex Instructions 1Document15 pagesVortex Instructions 1Anonymous xFDpeji3HWPas encore d'évaluation
- L Eau Et Le Principe de VieDocument39 pagesL Eau Et Le Principe de VieJean-Loïc BauchetPas encore d'évaluation
- DigestionDocument15 pagesDigestionkenniaevelynsullcabePas encore d'évaluation
- Kit de DynamisationDocument2 pagesKit de DynamisationJean-Loïc BauchetPas encore d'évaluation
- La Notion de Terrain BiologiqueDocument2 pagesLa Notion de Terrain BiologiqueJean-Loïc BauchetPas encore d'évaluation
- Le PH 1. Lautoprotolyse de Leau - 2014Document6 pagesLe PH 1. Lautoprotolyse de Leau - 2014MouadBastaouiPas encore d'évaluation
- Les Étapes de La Réaction InflammatoireDocument6 pagesLes Étapes de La Réaction InflammatoireJean-Loïc BauchetPas encore d'évaluation
- L Ensemble Arc-En-CielDocument35 pagesL Ensemble Arc-En-CielJean-Loïc BauchetPas encore d'évaluation
- La Mort Par La MedecineDocument10 pagesLa Mort Par La MedecineJean-Loïc BauchetPas encore d'évaluation
- Élément OxygeneDocument38 pagesÉlément OxygeneJean-Loïc BauchetPas encore d'évaluation
- Gerson KelleyDocument52 pagesGerson KelleyAssia Oumou MohamedPas encore d'évaluation
- Index Phytotherapie PDFDocument20 pagesIndex Phytotherapie PDFeforgues564100% (1)
- Heureux Chaque JourDocument111 pagesHeureux Chaque JourcenderillastoryPas encore d'évaluation
- Initiation À La Spagyrie Et Aux Médecines AlchimiquesDocument11 pagesInitiation À La Spagyrie Et Aux Médecines Alchimiquesphilosophe662511Pas encore d'évaluation
- EndofDocument7 pagesEndofOusmanePas encore d'évaluation
- HYDROGENE PDF OK Effets Bénéfiques - GEOENERGIES PagesDocument11 pagesHYDROGENE PDF OK Effets Bénéfiques - GEOENERGIES PagesJean-Loïc BauchetPas encore d'évaluation
- Grecs 4 ElementsDocument7 pagesGrecs 4 ElementsJean-Loïc BauchetPas encore d'évaluation
- DavenportDocument28 pagesDavenportCheick SANOUPas encore d'évaluation
- Fleurs de Bach PDFDocument38 pagesFleurs de Bach PDFNulli TasPas encore d'évaluation
- Élément OxygeneDocument38 pagesÉlément OxygeneJean-Loïc BauchetPas encore d'évaluation
- Equilibre-Acide-Base VIDocument33 pagesEquilibre-Acide-Base VIANDRIANOME tafitaPas encore d'évaluation
- EFFETS TAMPONS RÉGULATION DU PHDocument26 pagesEFFETS TAMPONS RÉGULATION DU PHJean-Loïc BauchetPas encore d'évaluation
- Manual Walther CP88 Letter FR (3119)Document9 pagesManual Walther CP88 Letter FR (3119)Alex SangokuPas encore d'évaluation
- BETON ARME Chap 3Document38 pagesBETON ARME Chap 3Qastali AbdeLlatifPas encore d'évaluation
- Les Adolescents Et Le SportDocument3 pagesLes Adolescents Et Le SportMassa DiamondPas encore d'évaluation
- Infection Bactérienne: Quelle Place Pour La Phagothérapie ? Bacterial Infection - Update On Phage TherapyDocument3 pagesInfection Bactérienne: Quelle Place Pour La Phagothérapie ? Bacterial Infection - Update On Phage TherapyM xPas encore d'évaluation
- GelatineDocument8 pagesGelatineonerPas encore d'évaluation
- Electro SecondeDocument72 pagesElectro SecondeNDAVKOUDA DZARAPas encore d'évaluation
- Gare À Gare À La Malbouff e ! La Malbouff e !: L'Algérie Améliore Son ClassementDocument28 pagesGare À Gare À La Malbouff e ! La Malbouff e !: L'Algérie Améliore Son ClassementMohand BakirPas encore d'évaluation
- Etude D'impact EnvironementalDocument27 pagesEtude D'impact Environementalmohammed amine qaouriPas encore d'évaluation
- Calcule de Cylindrée ProfDocument6 pagesCalcule de Cylindrée ProfLudo RenardPas encore d'évaluation
- Almaden Morocco FRDocument20 pagesAlmaden Morocco FRMakrani BrahimPas encore d'évaluation
- Metering Units b048 Bro FR Rev0Document16 pagesMetering Units b048 Bro FR Rev0Peter BirdPas encore d'évaluation
- Je M'interroge Sur Mon Exp Erience Des Tpe Math-SvtDocument10 pagesJe M'interroge Sur Mon Exp Erience Des Tpe Math-SvttboiPas encore d'évaluation
- Lettre D'engagements de Monsieur LEFORT NICOLASDocument3 pagesLettre D'engagements de Monsieur LEFORT NICOLASNico LynoPas encore d'évaluation
- UntitledDocument15 pagesUntitledValerie MONTEILPas encore d'évaluation
- Tca ExposeDocument38 pagesTca ExposeAmeh Kouadio67% (3)
- Neurosciences Cliniques. de La Perception Aux Troubles Du Comportement-2008Document440 pagesNeurosciences Cliniques. de La Perception Aux Troubles Du Comportement-2008Hanen KhiariPas encore d'évaluation
- IDEreanimationDocument106 pagesIDEreanimationderty100% (1)
- CALENDRIER AGRICOLE POUR LA CAMPAGNE 2020 (Enregistré Automatiquement)Document18 pagesCALENDRIER AGRICOLE POUR LA CAMPAGNE 2020 (Enregistré Automatiquement)Simon Pierre NGONO ETOGOPas encore d'évaluation
- Cours Uvi2 - ADocument69 pagesCours Uvi2 - AMarie Celine JulienPas encore d'évaluation
- RandrianavonyPatricia SN HDR 15Document114 pagesRandrianavonyPatricia SN HDR 15Fy EzahanaPas encore d'évaluation
- 4-Les Différents Types de Déformations - CopieDocument3 pages4-Les Différents Types de Déformations - CopieEyouta EyaPas encore d'évaluation