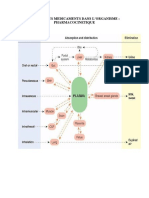
Vous aimerez peut-être aussi
- Cours Pharmacocinétique 2020Document74 pagesCours Pharmacocinétique 2020Lord GlorfindelPas encore d'évaluation
- Cinetique 2Document66 pagesCinetique 2Zeinebou AliounePas encore d'évaluation
- UE 2.3 Pharmacologie G N RaleDocument119 pagesUE 2.3 Pharmacologie G N RaleAbdel MonkouPas encore d'évaluation
- Pharmacocinétique Qualitative Et QuantitativeDocument66 pagesPharmacocinétique Qualitative Et QuantitativeAya SAIDIPas encore d'évaluation
- PharmacocinétiqueDocument94 pagesPharmacocinétiqueOMAR EL HAMDAOUIPas encore d'évaluation
- 1 - PharmacocinétiqueDocument18 pages1 - PharmacocinétiqueZaid YoussefPas encore d'évaluation
- Chapitre 2 Pharmacologie Bourouba S1Document10 pagesChapitre 2 Pharmacologie Bourouba S1Bel KisPas encore d'évaluation
- 5-PHARMACOCINETIQUE (Résorption) 2021Document32 pages5-PHARMACOCINETIQUE (Résorption) 2021Rania MaddahPas encore d'évaluation
- Pharmaco Cine TiqueDocument85 pagesPharmaco Cine TiqueAli AsdadiPas encore d'évaluation
- Chapitre I PharmacocinétiqueDocument5 pagesChapitre I PharmacocinétiqueDan NdiPas encore d'évaluation
- Stanke Labesque Francoise p02Document84 pagesStanke Labesque Francoise p02Jm NjmPas encore d'évaluation
- 5 - PharmacocinétiquesDocument3 pages5 - Pharmacocinétiquessamir hamadPas encore d'évaluation
- Pharmacocinetique QualitativeDocument45 pagesPharmacocinetique Qualitativecep100% (3)
- 3-Paramètres Pharmacocinétiques 3eme Année PharmacologieDocument3 pages3-Paramètres Pharmacocinétiques 3eme Année Pharmacologieraounek derradjiPas encore d'évaluation
- Pharmacologie GénéraleDocument50 pagesPharmacologie GénéralePascal LandsheerePas encore d'évaluation
- 2-Absorption DistributionDocument38 pages2-Absorption DistributionLaMri IkRamPas encore d'évaluation
- Pharmaco Cine TiqueDocument28 pagesPharmaco Cine TiqueANONYME179Pas encore d'évaluation
- Pharmacologie GeneraleDocument35 pagesPharmacologie GeneraleNesrine RAPas encore d'évaluation
- Chapitre 1 Pharmacologie Gle - 3Document30 pagesChapitre 1 Pharmacologie Gle - 3M.B. IsmailPas encore d'évaluation
- BiodisponibilitéDocument10 pagesBiodisponibilitéMaelys BressonPas encore d'évaluation
- 3 - Absorption Des MédicamentsDocument4 pages3 - Absorption Des Médicamentsazerty azertyPas encore d'évaluation
- Absorption Distribution 2023Document38 pagesAbsorption Distribution 2023Hiba HibaPas encore d'évaluation
- La Pharmacocinetique C Debraine PDFDocument72 pagesLa Pharmacocinetique C Debraine PDFMaria RPas encore d'évaluation
- Intelligentsia - Cours D'introduction de PharmacologieDocument21 pagesIntelligentsia - Cours D'introduction de PharmacologieGabin KenfackPas encore d'évaluation
- 1 - PharmacocinétiqueDocument4 pages1 - PharmacocinétiqueGillian BarnyPas encore d'évaluation
- Cours de Pharmacie Galénique La BiopharmacieDocument14 pagesCours de Pharmacie Galénique La BiopharmacieZineb fella Mahi100% (1)
- Pharmaco GénéraleDocument11 pagesPharmaco Généralesarah.ben.mtpPas encore d'évaluation
- Cours Controle QualitéDocument88 pagesCours Controle QualitéWalid Masmoudi100% (1)
- 1 11 Principes de Pharmacologie de Pharmacocinétique Et PharmacodynamieDocument7 pages1 11 Principes de Pharmacologie de Pharmacocinétique Et PharmacodynamieRim BenguenabPas encore d'évaluation
- PharmacocinétiqueDocument20 pagesPharmacocinétiquesherry1808Pas encore d'évaluation
- Abs-Dis Med Dent CHADOUDocument40 pagesAbs-Dis Med Dent CHADOUmakhlouf.farah02Pas encore d'évaluation
- Principes PharmacocinétiquesDocument124 pagesPrincipes PharmacocinétiquesKa Tia100% (1)
- Devenir Du Médicament Dans L'org Optic2023Document8 pagesDevenir Du Médicament Dans L'org Optic2023ikrambout55Pas encore d'évaluation
- Pharmacocinétique Et Métabolisme Des MédicamentsDocument7 pagesPharmacocinétique Et Métabolisme Des MédicamentsAbenekPas encore d'évaluation
- Chapitre 1 La PharmacocinétiqueDocument5 pagesChapitre 1 La PharmacocinétiquebilalPas encore d'évaluation
- Ch2-Biopharmacie GaléniqueDocument3 pagesCh2-Biopharmacie GaléniqueSouhila BennakhlaPas encore d'évaluation
- La BiodisponibilitéDocument5 pagesLa BiodisponibilitéYas Miine100% (1)
- PK 1Document39 pagesPK 1Ahmadou Dame GAYEPas encore d'évaluation
- Chapitre I Pharmacocinetique 2023Document89 pagesChapitre I Pharmacocinetique 2023Abderahmane KmrPas encore d'évaluation
- La Biopharmacie 2022Document5 pagesLa Biopharmacie 2022Chahinez BaPas encore d'évaluation
- 07 - PharmacocinétiqueDocument25 pages07 - PharmacocinétiqueAdam BennaniPas encore d'évaluation
- Pharmacologie 2020-Dr SoafaraDocument44 pagesPharmacologie 2020-Dr SoafaraFahendrenaPas encore d'évaluation
- Pharmacocinetique QuantitativeDocument29 pagesPharmacocinetique Quantitativecep100% (2)
- 2 PharmacocinétiqueDocument37 pages2 Pharmacocinétiqueʚï Sara ÏɞPas encore d'évaluation
- Toxicologie Générale - ToxicocinétiqueDocument6 pagesToxicologie Générale - ToxicocinétiqueAyoub BENSAKHRIA100% (1)
- ABSORBTIONDocument28 pagesABSORBTIONrima.lettreuchPas encore d'évaluation
- 01 Pharma Chapitre IDocument21 pages01 Pharma Chapitre IDavid MaynéPas encore d'évaluation
- Cours 1 PharmacologieDocument26 pagesCours 1 Pharmacologieannasicsic12Pas encore d'évaluation
- Pharm 30-11-2017Document19 pagesPharm 30-11-2017Elina GhochiPas encore d'évaluation
- Pharmacocinétique Limonade 2Document71 pagesPharmacocinétique Limonade 2Salomon JosephPas encore d'évaluation
- Toxicologie Et PharmacologieDocument12 pagesToxicologie Et PharmacologieYannick DsprbsPas encore d'évaluation
- Chap 1Document40 pagesChap 1NadinePas encore d'évaluation
- GaléniqueDocument16 pagesGaléniquePph Promo22Pas encore d'évaluation
- Notions de Pharmacologie 1Document20 pagesNotions de Pharmacologie 1Oumar TraoréPas encore d'évaluation
- Pharmaco3an16 PharmacocinetiqueDocument30 pagesPharmaco3an16 PharmacocinetiqueTr LwPas encore d'évaluation
- Cours Biopharmacie Et PharmacocinétiqueDocument56 pagesCours Biopharmacie Et PharmacocinétiqueSalah Sparrow75% (4)
- Concepts Généraux de PKDocument34 pagesConcepts Généraux de PKZitouni LaminePas encore d'évaluation
- Introduction À La Pharmacocinétique: Disponible Sur Le SiteDocument93 pagesIntroduction À La Pharmacocinétique: Disponible Sur Le SiteIssam mairfatPas encore d'évaluation
- Pharmacologie: Les Grands Articles d'UniversalisD'EverandPharmacologie: Les Grands Articles d'UniversalisPas encore d'évaluation
- Pose D'une Vessie de GlaceDocument13 pagesPose D'une Vessie de GlaceSaîda SalahPas encore d'évaluation
- Anatomie Physiologie Système Nerveux Résumé CourtDocument119 pagesAnatomie Physiologie Système Nerveux Résumé CourtSaîda SalahPas encore d'évaluation
- ENGAGEMENT Réglement ISPITS 2019-20Document1 pageENGAGEMENT Réglement ISPITS 2019-20Saîda SalahPas encore d'évaluation
- Pneumopathies Communautaires AiguesDocument39 pagesPneumopathies Communautaires AiguesSaîda SalahPas encore d'évaluation
- Grossesse Extra UterineDocument11 pagesGrossesse Extra UterineSaîda SalahPas encore d'évaluation
- Cours de Communication Mme OUKSAKADocument187 pagesCours de Communication Mme OUKSAKASaîda SalahPas encore d'évaluation
- Nutrition Regime AlimentaireDocument186 pagesNutrition Regime AlimentaireSaîda Salah100% (1)
- Les Structures D'urgenceDocument33 pagesLes Structures D'urgenceSaîda SalahPas encore d'évaluation
- MethodologieDocument5 pagesMethodologieSaîda SalahPas encore d'évaluation
- Grossesse Extra UterineDocument11 pagesGrossesse Extra UterineSaîda SalahPas encore d'évaluation
- Droits Et Devoirs de L'ideDocument36 pagesDroits Et Devoirs de L'ideMohamed InnekidenePas encore d'évaluation
- Les Formes À Libération ProlongéeDocument21 pagesLes Formes À Libération ProlongéeSilia ZemouliPas encore d'évaluation
- Pharmacologie A3Document18 pagesPharmacologie A3A100% (1)
- Atepadene PDFDocument2 pagesAtepadene PDFSérîîna LauretPas encore d'évaluation
- Formulaire de Demande Dagrement Dun Etablissement PharmaceutiqueDocument2 pagesFormulaire de Demande Dagrement Dun Etablissement PharmaceutiqueHaddadou HocinePas encore d'évaluation
- Caractérisation Physico-ChimiqueDocument81 pagesCaractérisation Physico-ChimiqueAmina BadaouiPas encore d'évaluation
- DM 4 SujetDocument4 pagesDM 4 SujetnicochocoPas encore d'évaluation
- Liste Médicaments ÉcrasablesDocument29 pagesListe Médicaments ÉcrasablesmeglinkyPas encore d'évaluation
- PREZTEBIMOSADocument20 pagesPREZTEBIMOSAEMFLYFILMS TVPas encore d'évaluation
- 2021 Repertoire Medicament Relevant Des Structures de L OSPES PDFDocument121 pages2021 Repertoire Medicament Relevant Des Structures de L OSPES PDFezzahmed2023Pas encore d'évaluation
- 1EMD 2016 Corrig Type 3anDocument6 pages1EMD 2016 Corrig Type 3anKhàoulà Đjm100% (1)
- Tableaux Comparatifs-MedicamentsDocument12 pagesTableaux Comparatifs-MedicamentsphilippePas encore d'évaluation
- Pharmacie GaleniqueDocument9 pagesPharmacie Galeniquemondher64Pas encore d'évaluation
- Décret N2007-157 - Subs VénéneusesDocument3 pagesDécret N2007-157 - Subs VénéneusesVERONIQUE SALESPas encore d'évaluation
- Attestation de Prise en - ChargeDocument1 pageAttestation de Prise en - ChargebenzakourPas encore d'évaluation
- DEUST 1 S1 5 Accueil Du Patient Sans ordoSEGUIN 2Document97 pagesDEUST 1 S1 5 Accueil Du Patient Sans ordoSEGUIN 2meriam.aodPas encore d'évaluation
- Livret Stage 6e AnnéeDocument15 pagesLivret Stage 6e AnnéeRIM ARHERBIPas encore d'évaluation
- TP SaraaDocument8 pagesTP SaraaYasmine YasminePas encore d'évaluation
- Coordonee Des Pharmacies 1Document26 pagesCoordonee Des Pharmacies 1Zo PatrickPas encore d'évaluation
- Pharmacovigilance Des ARVDocument12 pagesPharmacovigilance Des ARVAlly BPas encore d'évaluation
- Branches de La PharmacologieDocument8 pagesBranches de La PharmacologieradeyymvnprodPas encore d'évaluation
- Comprime Gelule SachetDocument20 pagesComprime Gelule SachetKomla Edem Dieudonné AdjonyoPas encore d'évaluation
- MethadoneDocument2 pagesMethadoneAya EssamriPas encore d'évaluation
- Planning DFASP1 - S7 - TP - TDDocument3 pagesPlanning DFASP1 - S7 - TP - TDazizbenchihaPas encore d'évaluation
- Formulation Transposition DechelleDocument15 pagesFormulation Transposition DechelleAbdel HebirPas encore d'évaluation