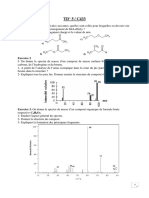
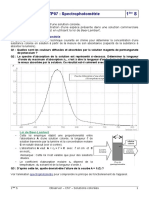
Vous aimerez peut-être aussi
- COURS-Spectroscopie IR-UVDocument28 pagesCOURS-Spectroscopie IR-UVHa NahPas encore d'évaluation
- Cour SpectroDocument33 pagesCour SpectroRadjaa AddPas encore d'évaluation
- Cours IR TableauxDocument27 pagesCours IR TableauxMojinPas encore d'évaluation
- Fiche de TD M1 Macromoléculaire, Propriétés Physico-Chimiques Des PolymèresDocument8 pagesFiche de TD M1 Macromoléculaire, Propriétés Physico-Chimiques Des PolymèresHanane DjaffarPas encore d'évaluation
- 3.transitions D'énergie Électroniques Et Vibratoires PDFDocument6 pages3.transitions D'énergie Électroniques Et Vibratoires PDFkimmikPas encore d'évaluation
- 2008 IR RMN CorrectionDocument17 pages2008 IR RMN CorrectionKadaPas encore d'évaluation
- Compte Rendu Du TP de RMN:: Réalisé Par: AMINA EL ALJ Sous-Direction De: HAMDOUN GHANEMDocument9 pagesCompte Rendu Du TP de RMN:: Réalisé Par: AMINA EL ALJ Sous-Direction De: HAMDOUN GHANEMamina elaljPas encore d'évaluation
- CHM 1302 - 2005 - Part 1 - GiassonDocument16 pagesCHM 1302 - 2005 - Part 1 - GiassonMiskine FilsPas encore d'évaluation
- TD Ir 2019Document39 pagesTD Ir 2019khouloud bensalemPas encore d'évaluation
- Cours Spectre de Masse - PR MAMOUNI - 2Document31 pagesCours Spectre de Masse - PR MAMOUNI - 2abd samadPas encore d'évaluation
- Partie Groupements Fonctionnels Cours Chimie Organique 2021Document32 pagesPartie Groupements Fonctionnels Cours Chimie Organique 2021Kamal Dehbi100% (1)
- Les Cellules Photovoltaïques Hybrides À Colorant: Institut Des Sciences Moléculaires - Université de BordeauxDocument26 pagesLes Cellules Photovoltaïques Hybrides À Colorant: Institut Des Sciences Moléculaires - Université de Bordeauxbouchra houdaPas encore d'évaluation
- TP FTIRDocument3 pagesTP FTIRMerry MerryPas encore d'évaluation
- Exer Spectro - Suppl Et CorrigeDocument34 pagesExer Spectro - Suppl Et CorrigeMichael Lee100% (1)
- Controle 3 - 1 S2 TC 19 - 20Document4 pagesControle 3 - 1 S2 TC 19 - 20Chaoui YoussefPas encore d'évaluation
- Molécule FuraneDocument11 pagesMolécule FuraneHamza BoulikaPas encore d'évaluation
- Série 1Document2 pagesSérie 1saidPas encore d'évaluation
- 3.spectre RMN Du Proton PDFDocument5 pages3.spectre RMN Du Proton PDFkimmikPas encore d'évaluation
- Table FTIRDocument5 pagesTable FTIRMohamed El Mehdi MEKHZOUMPas encore d'évaluation
- TP19-Spectroscopie IRDocument4 pagesTP19-Spectroscopie IRSouhila Benkhaled100% (1)
- Chapitre - III 20 05 2021Document34 pagesChapitre - III 20 05 2021Jhee raaPas encore d'évaluation
- Corrigé Des TD Méthodes Specrtoscopiques - Auto ÉvaluationDocument3 pagesCorrigé Des TD Méthodes Specrtoscopiques - Auto ÉvaluationSimo WacPas encore d'évaluation
- TD 3 - CorrigéDocument5 pagesTD 3 - CorrigéAbdelhamid ABDENNOURIPas encore d'évaluation
- LPro Spectro Picquet PDFDocument173 pagesLPro Spectro Picquet PDFJunior MbatepPas encore d'évaluation
- Série Cpg1.Document3 pagesSérie Cpg1.lahyaniPas encore d'évaluation
- TD° 5 C433 - CopieDocument3 pagesTD° 5 C433 - CopieRayene BARPas encore d'évaluation
- CHAPVDocument27 pagesCHAPVM LPas encore d'évaluation
- LPro ElectroDocument23 pagesLPro ElectroYoussef Akr100% (1)
- Cours Séance 1 DDSDocument14 pagesCours Séance 1 DDSMohammed REMAIDIPas encore d'évaluation
- Série Dexercices 2 - Spectrométrie Infra RougeDocument3 pagesSérie Dexercices 2 - Spectrométrie Infra RougeSamar ZedamPas encore d'évaluation
- Correction Série D'exercices 1 RMNDocument4 pagesCorrection Série D'exercices 1 RMNToulmoutine ManelPas encore d'évaluation
- RMNCours TDOct 2016Document2 pagesRMNCours TDOct 2016Cafe EstudiantinPas encore d'évaluation
- c06 Cours Spectres RMN 2Document4 pagesc06 Cours Spectres RMN 2Ganiyou AdenidjiPas encore d'évaluation
- Spectroscopie IR 3Document45 pagesSpectroscopie IR 3hind bioPas encore d'évaluation
- TP RMN 2Document9 pagesTP RMN 2Nour BkPas encore d'évaluation
- Chap1, Notioncinétique 1Document59 pagesChap1, Notioncinétique 1Safae RezzoukPas encore d'évaluation
- Tube À RXDocument13 pagesTube À RXzinebPas encore d'évaluation
- Huckel 1Document4 pagesHuckel 1Khalid ZegPas encore d'évaluation
- 5 Spectroscopie IRDocument98 pages5 Spectroscopie IRZora El BouloulPas encore d'évaluation
- Elemchim CC2 2014 Corr PDFDocument2 pagesElemchim CC2 2014 Corr PDFNour TaboubiPas encore d'évaluation
- Série 1 UVDocument3 pagesSérie 1 UVam.ayouazPas encore d'évaluation
- Chapitre IIIDocument11 pagesChapitre IIIAmadji YassminPas encore d'évaluation
- Les Liaisons ChimiquesDocument30 pagesLes Liaisons ChimiquesEmadPas encore d'évaluation
- 4 Orga 2 PDFDocument80 pages4 Orga 2 PDFKamel HachamaPas encore d'évaluation
- RMN 02Document87 pagesRMN 02Abderrahim BelmJouJPas encore d'évaluation
- 698rcp-Examen M32Document8 pages698rcp-Examen M329anbo3 TVPas encore d'évaluation
- Cinetique Des PolymereDocument51 pagesCinetique Des PolymerejakkkkPas encore d'évaluation
- Corrigé Examen 2017-2018Document4 pagesCorrigé Examen 2017-2018Leo Martinez100% (1)
- Correction Epreuve de Chimie Des Electrolytes 2014 2015Document5 pagesCorrection Epreuve de Chimie Des Electrolytes 2014 2015Imene GhmrPas encore d'évaluation
- Cours Chimie Organique SMP S3Document68 pagesCours Chimie Organique SMP S3MOHAMMED ZAKARIA BAALI100% (1)
- Chim430 Organometallique Cours PDFDocument144 pagesChim430 Organometallique Cours PDFAmel AyadiPas encore d'évaluation
- Cours Fluorescence - 1Document51 pagesCours Fluorescence - 1ezo ezoPas encore d'évaluation
- Spectroscopie RamanDocument7 pagesSpectroscopie RamanKenzaPas encore d'évaluation
- TS StereoisomerieDocument3 pagesTS StereoisomerieJeanne RousseauPas encore d'évaluation
- Cours RMN Proton 2021-ConvertiDocument11 pagesCours RMN Proton 2021-ConvertiKamer BourahlaPas encore d'évaluation
- Cours Et ExercicesDocument41 pagesCours Et ExercicesM.B. IsmailPas encore d'évaluation
- Cours Catalyse Par Les Complexe de Meteaux de TransitionDocument19 pagesCours Catalyse Par Les Complexe de Meteaux de TransitionMohamed EL FAGHLOUMI100% (1)
- TP de La Spectroscopie INFRA ROUGEDocument25 pagesTP de La Spectroscopie INFRA ROUGEAhmed Dris100% (1)
- Méthodes Spectroscopiques D'analyseDocument49 pagesMéthodes Spectroscopiques D'analyseRoumaissa LbPas encore d'évaluation
- Xerrada Banc JBB 2010Document32 pagesXerrada Banc JBB 2010fellah aminaPas encore d'évaluation
- Les ElectrophorèsesDocument31 pagesLes Electrophorèsesfellah aminaPas encore d'évaluation
- Myxomycète, Oomycètes ChitridiomycètesDocument33 pagesMyxomycète, Oomycètes Chitridiomycètesfellah aminaPas encore d'évaluation
- Legislation EnvirommentaleDocument39 pagesLegislation Envirommentalefellah aminaPas encore d'évaluation
- La Chromatographie Sur Couche Mince 1.principe: Phénomène D'élutionDocument6 pagesLa Chromatographie Sur Couche Mince 1.principe: Phénomène D'élutionfellah aminaPas encore d'évaluation
- I. Cycles Biogéochimiques Cycle Du CarboneDocument3 pagesI. Cycles Biogéochimiques Cycle Du Carbonefellah aminaPas encore d'évaluation
- IV. Fonctionnement Des ÉcosystèmesDocument7 pagesIV. Fonctionnement Des Écosystèmesfellah aminaPas encore d'évaluation
- INTRODUCTION, LES METABOLITES MICROBIENS 3 PagesDocument3 pagesINTRODUCTION, LES METABOLITES MICROBIENS 3 Pagesfellah aminaPas encore d'évaluation
- ClostridiumDocument36 pagesClostridiumfellah aminaPas encore d'évaluation
- Entérobac A Cours 1Document94 pagesEntérobac A Cours 1fellah aminaPas encore d'évaluation
- 3 Ème Cours Ecologie L2Document26 pages3 Ème Cours Ecologie L2fellah aminaPas encore d'évaluation
- Cocci À Gram Négatif by TOUATIDocument11 pagesCocci À Gram Négatif by TOUATIfellah aminaPas encore d'évaluation
- SUBSTRATS CARBONES INDUSTRIELS 8 PagesDocument8 pagesSUBSTRATS CARBONES INDUSTRIELS 8 Pagesfellah aminaPas encore d'évaluation
- Les Inocula IndustrielsDocument4 pagesLes Inocula IndustrielsmokranimaminePas encore d'évaluation
- Concours Doctorat LMD 2016-2017Document7 pagesConcours Doctorat LMD 2016-2017fellah aminaPas encore d'évaluation
- Interactions MicrobiennesDocument99 pagesInteractions Microbiennesfellah aminaPas encore d'évaluation
- Placard-Concours EPAU 2017-2018Document1 pagePlacard-Concours EPAU 2017-2018fellah aminaPas encore d'évaluation
- Résulats Finaux Du Concours de Doctorat en Ecologie Environnement, Spécialité BSA 2018 2019Document3 pagesRésulats Finaux Du Concours de Doctorat en Ecologie Environnement, Spécialité BSA 2018 2019fellah aminaPas encore d'évaluation
- EcologieDocument67 pagesEcologiefellah aminaPas encore d'évaluation
- Placard DLMD 2017-2018 - 2Document1 pagePlacard DLMD 2017-2018 - 2fellah aminaPas encore d'évaluation
- Ecologie GénéraleDocument23 pagesEcologie GénéraleNabil holmesPas encore d'évaluation
- COURS Gestion Des Espèces Animales 1Document2 pagesCOURS Gestion Des Espèces Animales 1fellah aminaPas encore d'évaluation
- Cours 1 Gestion de La FauneDocument61 pagesCours 1 Gestion de La Faunefellah aminaPas encore d'évaluation
- Cours 15 - MitochondriesDocument25 pagesCours 15 - Mitochondriesfellah aminaPas encore d'évaluation
- CytosqueletteDocument24 pagesCytosquelettefellah aminaPas encore d'évaluation
- Cours 4 - NoyauDocument11 pagesCours 4 - Noyaufellah aminaPas encore d'évaluation
- TD 2 GpcaDocument3 pagesTD 2 Gpcafellah aminaPas encore d'évaluation
- 7 Le Cytosquelette 6774c75e95Document29 pages7 Le Cytosquelette 6774c75e95fellah aminaPas encore d'évaluation
- 1 - Organisation Générale de La CelluleDocument7 pages1 - Organisation Générale de La Cellulefellah amina100% (1)
- Cours Chromato M1 BGVDocument8 pagesCours Chromato M1 BGVfouad elferdiPas encore d'évaluation
- 2-Appareillage Et Applications de l'UV (Partie 2)Document17 pages2-Appareillage Et Applications de l'UV (Partie 2)Maroua GuermatPas encore d'évaluation
- Spectrophotometrie MnO4Document4 pagesSpectrophotometrie MnO4Anaëlle Avril88% (8)
- Serie 10Document2 pagesSerie 10habessongPas encore d'évaluation
- COUPLAGE CG-SM-SM-p1487Document10 pagesCOUPLAGE CG-SM-SM-p1487salimPas encore d'évaluation
- Méthodes de SéparationDocument30 pagesMéthodes de SéparationNada MarhfourPas encore d'évaluation
- Serie 2 SMDocument7 pagesSerie 2 SMLINDA CHABANEPas encore d'évaluation
- CC1 Tas1Document2 pagesCC1 Tas1Kelli JinerPas encore d'évaluation
- Absorbance Et Loi de Beer-Lambert - Fiche de Révision - AnnabacDocument4 pagesAbsorbance Et Loi de Beer-Lambert - Fiche de Révision - AnnabacfatmamazharPas encore d'évaluation
- Utilization of Ion-Associate Formation in Conductimetric DeterminationDocument9 pagesUtilization of Ion-Associate Formation in Conductimetric DeterminationRaafat FarghalyPas encore d'évaluation
- Fichier Produit 2103Document5 pagesFichier Produit 2103wissou wissouPas encore d'évaluation
- Chromatographie Liquide de Haute Performance (HPLC)Document3 pagesChromatographie Liquide de Haute Performance (HPLC)Kahlouche HichemPas encore d'évaluation
- Spectroscopie Par Absorption Atomique Aas: Caracteristiques PrincipalesDocument2 pagesSpectroscopie Par Absorption Atomique Aas: Caracteristiques Principalesnadpharm13Pas encore d'évaluation
- HPLCDocument12 pagesHPLCimen GuizaniPas encore d'évaluation
- Dosage Par Etalonnage SpectrophotometriqueDocument3 pagesDosage Par Etalonnage Spectrophotometriqueelhidhab20140% (1)
- HPLCDocument5 pagesHPLCHadjer AdaidaPas encore d'évaluation
- P 41-46Document6 pagesP 41-46Nina PassiflowraPas encore d'évaluation
- TD N°3 Exercices D'application Sur Les Méthodes D'étude de La CelluleDocument2 pagesTD N°3 Exercices D'application Sur Les Méthodes D'étude de La CelluleMouhamed lamine SonkoPas encore d'évaluation
- TD 1Document2 pagesTD 1M L100% (1)
- Corrigé Des TD Méthodes Specrtoscopiques - Auto ÉvaluationDocument3 pagesCorrigé Des TD Méthodes Specrtoscopiques - Auto ÉvaluationSimo WacPas encore d'évaluation
- Exercices Corrigs de Spectroscopie de Masse PDFDocument2 pagesExercices Corrigs de Spectroscopie de Masse PDFTara70% (10)
- 14-11-18 1s4 TP Dakin CorrigeDocument3 pages14-11-18 1s4 TP Dakin CorrigeChrist AngePas encore d'évaluation
- Exposé Sur: PCR: Polymerase Chain ReactionDocument19 pagesExposé Sur: PCR: Polymerase Chain ReactionMallem SaPas encore d'évaluation
- Crs Bio Mol 2Document16 pagesCrs Bio Mol 2Fatima ChouatiPas encore d'évaluation
- TD 3 Fractionnement Celleulaire FinalDocument32 pagesTD 3 Fractionnement Celleulaire FinalAinganoun Mahcih100% (1)
- Spect Rosco PieDocument25 pagesSpect Rosco Pieadama nacanaboPas encore d'évaluation
- TD N°2 - Méthodes D'étude de La CelluleDocument4 pagesTD N°2 - Méthodes D'étude de La Cellulehidayet kdmPas encore d'évaluation
- HistoriqueDocument16 pagesHistoriqueImad Eddine ChaichiPas encore d'évaluation
- LPVPT - TD N°1 - TD N°2Document30 pagesLPVPT - TD N°1 - TD N°2Mimi BCGPas encore d'évaluation
- Série3 ChromatographieDocument6 pagesSérie3 ChromatographieIymen Bouti100% (1)
- Secrets ancestraux d'un maître guérisseur: Un sceptique occidental, un maître oriental et les plus grands secrets de la vieD'EverandSecrets ancestraux d'un maître guérisseur: Un sceptique occidental, un maître oriental et les plus grands secrets de la vieÉvaluation : 5 sur 5 étoiles5/5 (2)
- La vie des abeilles: Prix Nobel de littératureD'EverandLa vie des abeilles: Prix Nobel de littératureÉvaluation : 4 sur 5 étoiles4/5 (41)
- Harmonisation Energétique des Lieux: Habitat et haut-lieux sacrés 2020D'EverandHarmonisation Energétique des Lieux: Habitat et haut-lieux sacrés 2020Évaluation : 2.5 sur 5 étoiles2.5/5 (3)
- Améliorer votre mémoire: Un Guide pour l'augmentation de la puissance du cerveau, utilisant des techniques et méthodesD'EverandAméliorer votre mémoire: Un Guide pour l'augmentation de la puissance du cerveau, utilisant des techniques et méthodesÉvaluation : 5 sur 5 étoiles5/5 (2)
- L'Art de la guerre: Traité de stratégie en 13 chapitres (texte intégral)D'EverandL'Art de la guerre: Traité de stratégie en 13 chapitres (texte intégral)Évaluation : 4 sur 5 étoiles4/5 (3032)
- Électrotechnique | Pas à Pas: Bases, composants & circuits expliqués pour les débutantsD'EverandÉlectrotechnique | Pas à Pas: Bases, composants & circuits expliqués pour les débutantsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Manuel de fabrication du savon: Je fabrique mes savons facilementD'EverandManuel de fabrication du savon: Je fabrique mes savons facilementÉvaluation : 5 sur 5 étoiles5/5 (4)
- Transformez votre vie: Utilisez le pouvoir créateur qui est en vous pour construire votre vie à l'image de ce que vous voulez qu'elle soitD'EverandTransformez votre vie: Utilisez le pouvoir créateur qui est en vous pour construire votre vie à l'image de ce que vous voulez qu'elle soitÉvaluation : 4 sur 5 étoiles4/5 (14)
- Anatomie & 100 étirements essentiels: Techniques, Bénéfices attendus, Précautions à prendre, Conseils, Tableaux de séries, DouleursD'EverandAnatomie & 100 étirements essentiels: Techniques, Bénéfices attendus, Précautions à prendre, Conseils, Tableaux de séries, DouleursPas encore d'évaluation
- 160 ressources pour se lancer dans la vidéo quand on n’y connait rienD'Everand160 ressources pour se lancer dans la vidéo quand on n’y connait rienPas encore d'évaluation
- Cahier de jeux de stimulation cognitive: Sujets Alzheimer, désorientés, démences, amnésiesD'EverandCahier de jeux de stimulation cognitive: Sujets Alzheimer, désorientés, démences, amnésiesPas encore d'évaluation
- Enseigner une Langue Etrangère Par l’Apprentissage HybrideD'EverandEnseigner une Langue Etrangère Par l’Apprentissage HybridePas encore d'évaluation
- Approvisionnement et traitement de l’eau: Les Grands Articles d'UniversalisD'EverandApprovisionnement et traitement de l’eau: Les Grands Articles d'UniversalisPas encore d'évaluation
- Cancer - Leucémie: Et autres maladies apparemment incurables, mais guérissables avec des moyens naturelsD'EverandCancer - Leucémie: Et autres maladies apparemment incurables, mais guérissables avec des moyens naturelsPas encore d'évaluation
- 20 Véritables remèdes de nos grands-mères pour maigrir vite et enfin perdre du poidsD'Everand20 Véritables remèdes de nos grands-mères pour maigrir vite et enfin perdre du poidsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Puissance naturelles - Que faire si votre "meilleure partie" est en grève?: Puissance naturelles-améliorer les recours pour augmenter la virilité de la capacité à obtenir une érection de fermetéD'EverandPuissance naturelles - Que faire si votre "meilleure partie" est en grève?: Puissance naturelles-améliorer les recours pour augmenter la virilité de la capacité à obtenir une érection de fermetéÉvaluation : 3.5 sur 5 étoiles3.5/5 (3)
- Semer avec succès pour rassembler avec abundance. Jardin organique et synergique: Calcul des meilleurs jours pour l'ensemencement de chaque légumeD'EverandSemer avec succès pour rassembler avec abundance. Jardin organique et synergique: Calcul des meilleurs jours pour l'ensemencement de chaque légumePas encore d'évaluation
- Le B.A.-Ba de la communication: Comment convaincre, informer, séduire ?D'EverandLe B.A.-Ba de la communication: Comment convaincre, informer, séduire ?Évaluation : 3 sur 5 étoiles3/5 (1)
- Physique quantique pour les débutants: Découvrez les fondements de la mécanique quantique et la façon dont elle affecte le monde dans lequel nous vivons à travers ses théories les plus célèbresD'EverandPhysique quantique pour les débutants: Découvrez les fondements de la mécanique quantique et la façon dont elle affecte le monde dans lequel nous vivons à travers ses théories les plus célèbresÉvaluation : 5 sur 5 étoiles5/5 (2)
- Anatomie & 100 étirements essentiels pour le running: Principes de base, Techniques, Tableaux de séries, Précautions à prendre, Conseils, Programmes d'étirementsD'EverandAnatomie & 100 étirements essentiels pour le running: Principes de base, Techniques, Tableaux de séries, Précautions à prendre, Conseils, Programmes d'étirementsPas encore d'évaluation
- L'Art de La Magie au Bougie Wicca: Le Guide du Débutant à la Pratique de la Magie au Bougie de WiccaD'EverandL'Art de La Magie au Bougie Wicca: Le Guide du Débutant à la Pratique de la Magie au Bougie de WiccaÉvaluation : 3 sur 5 étoiles3/5 (1)
- Conception & Modélisation CAO: Le guide ultime du débutantD'EverandConception & Modélisation CAO: Le guide ultime du débutantPas encore d'évaluation
- Manuel pour les débutants Fabriquez des savons naturelsD'EverandManuel pour les débutants Fabriquez des savons naturelsÉvaluation : 3 sur 5 étoiles3/5 (2)
- La pensée dirigée: Traité sur le raisonnement et les logiquesD'EverandLa pensée dirigée: Traité sur le raisonnement et les logiquesÉvaluation : 5 sur 5 étoiles5/5 (2)