Vous aimerez peut-être aussi
- Hématopoïèse NSMDocument5 pagesHématopoïèse NSMMohamed ECHAMAIPas encore d'évaluation
- 1 - Hématopoïèse (Diapo)Document33 pages1 - Hématopoïèse (Diapo)Sara SouPas encore d'évaluation
- HématopoïèseDocument11 pagesHématopoïèseMohamed Amine BakariPas encore d'évaluation
- HématologieDocument58 pagesHématologieOUMAIMA EL CHAALPas encore d'évaluation
- Hémato CellulaireDocument20 pagesHémato CellulaireEl HanaPas encore d'évaluation
- Mega Cary Poi EseDocument33 pagesMega Cary Poi EseTaoufik Ben HoumichPas encore d'évaluation
- Hematopoiese Des Cellules SanguinesDocument29 pagesHematopoiese Des Cellules SanguinesedrylleelbartoPas encore d'évaluation
- 01 Granulopoiese Neutrophile Et Lignees Apparentees 1Document58 pages01 Granulopoiese Neutrophile Et Lignees Apparentees 1Hasan KaranbashPas encore d'évaluation
- Synthese HematologieDocument57 pagesSynthese HematologiemacfunzPas encore d'évaluation
- Hématopoïèse Couronné SocleDocument72 pagesHématopoïèse Couronné SocleSalam SawadogoPas encore d'évaluation
- 1) Diapo Pre-Requis-En-HematologieDocument54 pages1) Diapo Pre-Requis-En-HematologieEllive Eve50% (2)
- Fichier Produit 3693Document27 pagesFichier Produit 3693yaya camaraPas encore d'évaluation
- PR Belmekki GrannuloDocument56 pagesPR Belmekki Grannuloanaischauvin12Pas encore d'évaluation
- PPT Le Tissu Sanguin - PR AmegborDocument64 pagesPPT Le Tissu Sanguin - PR AmegborThéophile NAMOINEPas encore d'évaluation
- HématopoïèseDocument10 pagesHématopoïèseFayad BouraimaPas encore d'évaluation
- Hematopieza - Francezi-C2Document60 pagesHematopieza - Francezi-C2Buffy SundayPas encore d'évaluation
- Hématopoièse1 PDFDocument4 pagesHématopoièse1 PDFSarra BoutobzaPas encore d'évaluation
- Biologie AgadirDocument136 pagesBiologie Agadircimo.ouhammouPas encore d'évaluation
- Immunologie Appliquée USTHBDocument8 pagesImmunologie Appliquée USTHBmiliiiPas encore d'évaluation
- Hematopoiese 2013Document63 pagesHematopoiese 2013Njato FitahianahsPas encore d'évaluation
- Cours 1 Hematologie CellulaireDocument57 pagesCours 1 Hematologie CellulaireMohamed OuHouissPas encore d'évaluation
- 01 - Granulopoïèse Neutrophile Et Lignées ApparentéesDocument58 pages01 - Granulopoïèse Neutrophile Et Lignées ApparentéesSouhaib.el100% (1)
- 1 - H ©matopo Ø ®se 2020 FCBDocument59 pages1 - H ©matopo Ø ®se 2020 FCBsikaokoumassouPas encore d'évaluation
- Physiologie Du Sang G N Ralit DR - Bouchiha .PDF Filename Utf-8physiologie Du Sang Généralité DR - BouchihaDocument35 pagesPhysiologie Du Sang G N Ralit DR - Bouchiha .PDF Filename Utf-8physiologie Du Sang Généralité DR - BouchihaSalam SawadogoPas encore d'évaluation
- CARCINOGENESEDocument29 pagesCARCINOGENESEMĕ RïemPas encore d'évaluation
- Globules BlancsDocument65 pagesGlobules BlancsDestiny KoffiPas encore d'évaluation
- TD BiocellDocument16 pagesTD Biocelln hv hvhPas encore d'évaluation
- La Lignee GranuleuseDocument17 pagesLa Lignee Granuleuseyaya camaraPas encore d'évaluation
- Chapitre 1Document50 pagesChapitre 1Fella MazPas encore d'évaluation
- Globules Blancs PR YAODocument63 pagesGlobules Blancs PR YAOJean Luc Konan100% (1)
- TD2 - Cellules Souches, HeÌ Matopoiì Eì Se Exemples de Cancers Du SangDocument68 pagesTD2 - Cellules Souches, HeÌ Matopoiì Eì Se Exemples de Cancers Du Sangn hv hvhPas encore d'évaluation
- HématopoieseDocument21 pagesHématopoieseyaya camaraPas encore d'évaluation
- Syndrome MyéloproliferatifDocument74 pagesSyndrome MyéloproliferatifAli MoudPas encore d'évaluation
- Tissu Sanguin Pearl ComlanDocument58 pagesTissu Sanguin Pearl Comlansehoemmanuel15Pas encore d'évaluation
- 14-Pathologie-tumoraleDocument13 pages14-Pathologie-tumoraledanger inflammablePas encore d'évaluation
- Cours Hematologie CompletDocument337 pagesCours Hematologie CompletmukambafidelPas encore d'évaluation
- Generalites Sur L'hematologieDocument7 pagesGeneralites Sur L'hematologieYingra BaraPas encore d'évaluation
- 5b Thrombopã©niesDocument21 pages5b Thrombopã©niesLidvina FirquetPas encore d'évaluation
- Anapath Tumorale QEDocument5 pagesAnapath Tumorale QEDriss ElasriPas encore d'évaluation
- Granulopoïèse Et Sa RégulationDocument24 pagesGranulopoïèse Et Sa RégulationMor Ngom100% (1)
- Red B-Clin AmélioréeDocument27 pagesRed B-Clin AmélioréeEmmanuelPas encore d'évaluation
- 23-TUMEURS DU SYSTÈME NERVEUXDocument7 pages23-TUMEURS DU SYSTÈME NERVEUXdanger inflammablePas encore d'évaluation
- TP CytogénétiqueDocument92 pagesTP CytogénétiqueMouad HiliaPas encore d'évaluation
- 03 - Carcinogenèse - Réorganisation 23-24Document8 pages03 - Carcinogenèse - Réorganisation 23-24zinebtalbi213Pas encore d'évaluation
- Cytogénétique HumaineDocument128 pagesCytogénétique HumaineMohamed ECHAMAIPas encore d'évaluation
- H 2 PDFDocument2 pagesH 2 PDFAbdelillah AdamiPas encore d'évaluation
- Chromosome Et Caryotype (+ACC)Document13 pagesChromosome Et Caryotype (+ACC)sun-nee-chan9Pas encore d'évaluation
- MITOCHONDRIOPATHIESDocument51 pagesMITOCHONDRIOPATHIESLOKO Maoudi PaulettePas encore d'évaluation
- UE 2.2. Le SangDocument5 pagesUE 2.2. Le Sangaxel.lassagne26Pas encore d'évaluation
- 1 HematimetrieDocument11 pages1 HematimetriekhiralouibiPas encore d'évaluation
- 6-Les Populations Cellulaires LibresDocument50 pages6-Les Populations Cellulaires LibresAy ManPas encore d'évaluation
- 15 - Mécanisme de La CarcinogénèseDocument49 pages15 - Mécanisme de La CarcinogénèseManel Bouarbi100% (1)
- Cellule Souche HématopoietiqueDocument38 pagesCellule Souche HématopoietiqueYingra BaraPas encore d'évaluation
- Fichier Produit 1084Document17 pagesFichier Produit 1084Salomon JosephPas encore d'évaluation
- Cours de LhématopoïèseDocument15 pagesCours de LhématopoïèseIkbel WederniPas encore d'évaluation
- 4.tissu CancereuxDocument4 pages4.tissu Cancereuxroza jhjchvPas encore d'évaluation
- Hémato Léa-ArmandDocument8 pagesHémato Léa-Armandfreddo.chloePas encore d'évaluation
- 5) Formule LeucocytaireDocument43 pages5) Formule LeucocytaireFarida SirimaPas encore d'évaluation
- Surrénales: Les Grands Articles d'UniversalisD'EverandSurrénales: Les Grands Articles d'UniversalisPas encore d'évaluation
- Réponses Aux Questions À Propos de La BactériologieDocument2 pagesRéponses Aux Questions À Propos de La BactériologieMohamed ECHAMAIPas encore d'évaluation
- Six Sigma - OorekaDocument3 pagesSix Sigma - OorekaMohamed ECHAMAIPas encore d'évaluation
- Vaisseaux Du CoeurDocument2 pagesVaisseaux Du CoeurMohamed ECHAMAIPas encore d'évaluation
- Cytogénétique HumaineDocument128 pagesCytogénétique HumaineMohamed ECHAMAIPas encore d'évaluation
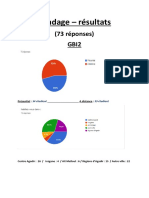
- Sondage Gbi2Document1 pageSondage Gbi2Mohamed ECHAMAIPas encore d'évaluation
- Ch1 - AtomistiqueDocument3 pagesCh1 - AtomistiqueMohamed ECHAMAIPas encore d'évaluation
- Les Peptides BenchakrounDocument90 pagesLes Peptides BenchakrounMohamed ECHAMAIPas encore d'évaluation
- Innervation Du CœurDocument1 pageInnervation Du CœurMohamed ECHAMAIPas encore d'évaluation
- Chimie 2 PDFDocument8 pagesChimie 2 PDFTarik Mlilas50% (2)
- Isa 2020Document1 pageIsa 2020Mohamed ECHAMAIPas encore d'évaluation
- Rapport Final Fermentation2Document63 pagesRapport Final Fermentation2Tor La sagesse100% (1)
- Les Matrices CoursDocument7 pagesLes Matrices CoursYassine MbzPas encore d'évaluation
- Aorte ThoraciqueDocument17 pagesAorte ThoraciqueMohamed ECHAMAIPas encore d'évaluation
- Notes CoeurDocument10 pagesNotes CoeurMohamed ECHAMAIPas encore d'évaluation
- Etude de La Diversité Métabolique Des Micro-Organismes Des Sources Hydrothermales OcéaniquesDocument206 pagesEtude de La Diversité Métabolique Des Micro-Organismes Des Sources Hydrothermales OcéaniquesMohamed ECHAMAIPas encore d'évaluation
- E Commerce Le Rapport (Version Finale)Document28 pagesE Commerce Le Rapport (Version Finale)pfe100% (1)
- Corrige 472768Document100 pagesCorrige 472768Kenz L'AïdPas encore d'évaluation
- Isa 2020Document1 pageIsa 2020Mohamed ECHAMAIPas encore d'évaluation
- Metabolisme Et Nutrition BacteriensDocument22 pagesMetabolisme Et Nutrition BacteriensMohamed ECHAMAIPas encore d'évaluation
- Compte RenduDocument12 pagesCompte RenduMohamed ECHAMAIPas encore d'évaluation
- La Culture in Vitro Astuces Et Pratiques: November 2015Document43 pagesLa Culture in Vitro Astuces Et Pratiques: November 2015Mohamed ECHAMAIPas encore d'évaluation
- Iaa (Oral)Document2 pagesIaa (Oral)Mohamed ECHAMAIPas encore d'évaluation
- Isa 2020Document1 pageIsa 2020Mohamed ECHAMAIPas encore d'évaluation
- Compte RenduDocument12 pagesCompte RenduMohamed ECHAMAIPas encore d'évaluation
- Enzymologie 9Document31 pagesEnzymologie 9Dr.Zakaria MFTPas encore d'évaluation
- Université Hassan II de Casablanca - Faculté Des Sciences Et Techniques de MohammediaDocument1 pageUniversité Hassan II de Casablanca - Faculté Des Sciences Et Techniques de MohammediaMohamed ECHAMAIPas encore d'évaluation
- Rapport SFE Les Huileries Du Souss BelhassanDocument38 pagesRapport SFE Les Huileries Du Souss BelhassanMohamed ECHAMAI100% (1)
- Le YaourtDocument21 pagesLe Yaourtcallm9100% (5)
- INTRODUCTIONDocument1 pageINTRODUCTIONMohamed ECHAMAIPas encore d'évaluation
- IV.7 - Infections Et GrossesseDocument7 pagesIV.7 - Infections Et GrossesseIman EL FEKAKPas encore d'évaluation
- Phytothrapiedesmaladiesdelappareildigestifa Kinshasa DRCongodocxDocument16 pagesPhytothrapiedesmaladiesdelappareildigestifa Kinshasa DRCongodocxØüssÂmāPas encore d'évaluation
- Santé Dentaire CommunautaireDocument6 pagesSanté Dentaire Communautairemerz RimPas encore d'évaluation
- MiliaireDocument7 pagesMiliaireAbd FaresPas encore d'évaluation
- Exercice Sur Les Procédés ExplicatifsDocument7 pagesExercice Sur Les Procédés Explicatifsfransec371% (21)
- David Icke 7 - 04 - 2020Document46 pagesDavid Icke 7 - 04 - 2020panasonic92Pas encore d'évaluation
- Notice Xydol 600mg Comp. Pelli. B 30Document1 pageNotice Xydol 600mg Comp. Pelli. B 30Mohamed Amine BouzianiPas encore d'évaluation
- dc2 4ème Sc-Exp 2022-2023Document3 pagesdc2 4ème Sc-Exp 2022-2023meriem zarrouk0% (1)
- Dent - AcupunctureDocument6 pagesDent - Acupunctureophil74Pas encore d'évaluation
- 5-Méthodes Dénombrement Et Identification - H LesouhaitierDocument15 pages5-Méthodes Dénombrement Et Identification - H LesouhaitierImanouila SilverPas encore d'évaluation
- Voie de Fait Nice Centre de LoisirDocument2 pagesVoie de Fait Nice Centre de LoisirFrance BleuPas encore d'évaluation
- 2 Terminologie Buemi PDFDocument30 pages2 Terminologie Buemi PDFmastermv2013Pas encore d'évaluation
- Synthèse Embryo 2005-2006Document35 pagesSynthèse Embryo 2005-2006Benjamin TchetcheroffPas encore d'évaluation
- Dzexams 1am Francais 365796Document4 pagesDzexams 1am Francais 365796new newPas encore d'évaluation
- QCMDocument74 pagesQCMNawel Lhk100% (2)
- Produits Dangereux 1Document9 pagesProduits Dangereux 1stifmarkos100% (1)
- Burn OutDocument41 pagesBurn OutingridguerreroocampoPas encore d'évaluation
- 00 Annales Zeros BacS 2 2 Spe Exemples1 Et 2Document7 pages00 Annales Zeros BacS 2 2 Spe Exemples1 Et 2Roland DjatsouPas encore d'évaluation
- Extrait Taher Ben JlounDocument22 pagesExtrait Taher Ben JlountehaPas encore d'évaluation
- Examen Blanc FMT 2020 - J2Document36 pagesExamen Blanc FMT 2020 - J2Abidi HichemPas encore d'évaluation
- Vitamine CDocument23 pagesVitamine CZaordoz ZedPas encore d'évaluation
- Soins Infirmiers MED1-DeN1Document100 pagesSoins Infirmiers MED1-DeN1Nelly OssangaPas encore d'évaluation
- 37 Rapport de Stage - Analyse Système Référence Et Con SavannesDocument52 pages37 Rapport de Stage - Analyse Système Référence Et Con SavannesÉric Comlan SewaiPas encore d'évaluation
- 007 Droit Du PatientDocument4 pages007 Droit Du PatientbrgPas encore d'évaluation
- Recos Antisepsie SF2H 2016Document92 pagesRecos Antisepsie SF2H 2016a k bPas encore d'évaluation
- Utérus Bicorne (DR Olivier Mukuku)Document5 pagesUtérus Bicorne (DR Olivier Mukuku)Docteur Olivier MukukuPas encore d'évaluation
- Elevation de La CreatininemieDocument12 pagesElevation de La CreatininemieNJEBARIKANUYE EugènePas encore d'évaluation
- Danièle Festy-Les Huiles Essentielles, Ça Marche !-LEDUC S. (2015)Document262 pagesDanièle Festy-Les Huiles Essentielles, Ça Marche !-LEDUC S. (2015)Hind Esserhani100% (1)
- 06 Memoire ProceduraleDocument10 pages06 Memoire ProceduraleLaetitia PicardPas encore d'évaluation
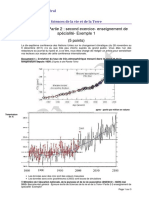
- Bac S 2018 SVT SpécialitéDocument4 pagesBac S 2018 SVT SpécialitéLETUDIANTPas encore d'évaluation